
Abstract
Background and Objectives
MCLA-128 is a bispecific monoclonal antibody targeting the HER2 and HER3 receptors and is in development to overcome HER3-mediated resistance to anti-HER2 therapies. The aims of this analysis were to characterize the population pharmacokinetics of MCLA-128 in patients with various solid tumors, to evaluate patient-related factors that affect the disposition of MCLA-128, and to assess whether flat dosing is appropriate.
Methods
MCLA-128 concentration data following intravenous administration were collected in a phase I/II clinical trial. Pharmacokinetic data were analyzed using non-linear mixed-effects modeling. Different compartmental models were evaluated. Various body size parameters including body weight, body surface area, and fat-free mass were evaluated as covariates in addition to age, sex, HER2 status, and tumor burden.
Results
In total, 1115 serum concentration measurements were available from 116 patients. The pharmacokinetics of MCLA-128 was best described by a two-compartment model with linear and non-linear (Michaelis–Menten) clearance. Fat-free mass significantly affected the linear clearance and volume of distribution of the central compartment of MCLA-128, explaining 8.4% and 5.6% of inter-individual variability, respectively. Tumor burden significantly affected the non-linear clearance capacity. Simulations demonstrated that dosing based on body size parameters resulted in similar area under the plasma concentration-time curve for a dosing interval (AUC0–τ), maximum and trough concentrations of MCLA-128, compared to flat dosing.
Conclusions
This analysis demonstrated that the pharmacokinetics of MCLA-128 exhibits similar disposition characteristics to other therapeutic monoclonal antibodies and that a flat dose of MCLA-128 in patients with various solid tumors is appropriate.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
MCLA-128 is a bispecific monoclonal antibody targeting HER2 and HER3 receptors and was evaluated in a phase I/II clinical trial in patients with various solid tumors. |
Pharmacokinetics of MCLA-128 were described by a two-compartment model with linear and non-linear clearance and fat-free mass was identified to significantly affect clearance and volume of distribution of the central compartment. |
Tumor burden was identified to significantly affect non-linear clearance capacity of MCLA-128. |
Regardless of the fat-free mass effect on the disposition of MCLA-128, flat dosing of MCLA-128 is appropriate in patients with solid tumors. |
A previously published preclinical pharmacokinetic model for MCLA-128 was able to predict clinical exposure to MCLA-128. |
1 Introduction
MCLA-128 is a full-length, humanized, immunoglobulin G1-bispecific monoclonal antibody with enhanced antibody-dependent cell-mediated cytotoxicity (ADCC). It targets the HER2 and HER3 transmembrane receptor tyrosine kinases. The mechanism of action is expected to rely on direct inhibition of tumor growth by blocking HER2:HER3 signaling and, via ADCC leading to elimination of tumor cells via recruitment of immune effector cells to tumor cells that have bound MCLA-128 [1, 2]. MCLA-128 is developed to overcome HER3-mediated resistance to epidermal growth factor receptor and HER2-targeted therapies in patients with HER2-overexpressing or HER2-amplified tumors.
Current HER2-targeted therapies are approved for HER2-amplified breast and gastric cancers, either as a single agent or in combination with other anticancer drugs [3, 4]. A proportion of patients treated with these therapies, however, show primary or acquired resistance [5, 6]. Resistance is often mediated by HER3 activation, either by upregulation of HER3 receptors in HER2-amplified tumors, or directly by the HER3 ligand, heregulin. Upregulation of HER3 in HER2-amplified tumors can result in ligand-independent dimerization of HER3 with HER2 and enhanced cell survival [7]. Alternatively, heregulin, drives dimerization of HER3 with HER2, resulting in potent activation of the PI3K/AKT pathway leading to enhanced growth and survival of HER2-amplified tumors. Heregulin stimulation was shown to mediate resistance to trastuzumab and lapatinib therapy [7,8,9]. The targeting of HER2 and HER3 by MCLA-128 could overcome this resistance. In vitro results have shown that MCLA-128 inhibits proliferation of HER2 over-expressing and HER2-low cells stimulated with heregulin. It also shows significantly higher potency than lapatinib, trastuzumab alone, or the combination of trastuzumab and pertuzumab [1, 2]. In vivo MCLA-128 demonstrates potent anti-tumor activity in relevant xenograft models [10]. Previously, the pharmacokinetics of MCLA-128 in cynomolgus monkeys were described by a two-compartment model with linear and non-linear clearance from the central compartment [10].
The objectives of this analysis are to characterize the population pharmacokinetics of MCLA-128 in patients with various solid tumors included in a phase I/II trial, identify patient-related factors that potentially influence the disposition of MCLA-128, and evaluate whether flat dosing of MCLA-128 would be appropriate. The first-in-human starting dose of MCLA-128 was partly based on simulations performed using the preclinical pharmacokinetic model. Therefore, the appropriateness of animal-to-human pharmacokinetic scaling was evaluated for MCLA-128.
2 Materials and Methods
2.1 Generation of MCLA-128
MCLA-128 was engineered using proprietary CH3 technology, and comprises two identical common light chains and two different heavy chains (anti-HER2 and anti-HER3). ADCC enhancement was achieved by low fucose glycoengineering using the GlymaxX® technology [2]. MCLA-128 is added to a saline solution before administration.
2.2 Data
This analysis was performed on data from patients in a phase I/II clinical trial (NCT02912949) of MCLA-128. The protocol was approved by the ethics committees of seven participating centers and all patients provided written informed consent before study entry. Data were pooled from the dose-escalation and dose-expansion cohorts. Patients in the dose-escalation cohorts were treated with flat doses ranging between 40 mg and 900 mg of MCLA-128, administered every 3 weeks (q3wk) and patients in the dose-expansion cohort were treated with a flat dose of 750 mg of MCLA-128, administered q3wk. MCLA-128 was administered intravenously as a 1-h (dose levels ≤ 360 mg) or 2-h infusion (dose levels > 360 mg). Patients with advanced solid tumors were included in the dose-escalation cohorts. Patients with selected advanced solid tumors (HER2 + gastric, HER2 + breast, endometrium, esophagus-gastric junction, colon, and non-small-cell lung carcinoma) were included in the dose-expansion cohort.
For the assessment of MCLA-128 pharmacokinetics, serum samples were collected on day 1 of cycle 1 at pre-dose, end of infusion, and 1, 2, 4, 8, and 24 h after end of infusion, any time on day 3 or 4, 8 and 15, and on day 1 of cycle 2, 3, and 4 at pre-dose and end of infusion. Samples were shipped frozen on dry ice and stored at − 80 °C until analysis. MCLA-128 was quantified in serum using a validated electrochemiluminescence immunoassay, with a lower limit of quantification (LLOQ) of 50 ng/mL. The intra- and inter-assay precision and accuracy of the method were within the target limits of 25% at the lower limit of quantification and within 20% for all other concentrations. Selectivity of MCLA-128 from human serum was reproducible and acceptable.
2.3 Population Pharmacokinetic Analysis
2.3.1 Structural Model
Non-linear mixed-effects modeling was used for the population pharmacokinetic analysis. The preclinical modeling analysis in cynomolgus monkeys identified a two-compartment model with linear and non-linear (Michaelis–Menten) clearance from the central compartment to best describe pharmacokinetic data of MCLA-128 [10]. Such model structures have been well established for the pharmacokinetics of other monoclonal antibodies as well, where the linear clearance describes FcRn-mediated clearance of monoclonal antibodies and the non-linear clearance target-mediated drug disposition [11]. Therefore, a two-compartment model was used as a starting point for the structural model building of the population pharmacokinetic analysis and the following parameters were estimated: linear clearance (CL), intercompartmental clearance (Q), volumes of distribution of the central and peripheral compartment (V1 and V2, respectively), the maximum elimination rate of the non-linear clearance (Vmax), and the Michaelis–Menten constant (Km). One- and two-compartment models with only linear clearance were also evaluated.
2.3.2 Statistical Model
Inter-individual variability (IIV) was evaluated for each of the pharmacokinetic parameters using an exponential model:
where \(P_{i}\) is the individual parameter estimate for individual \(i\), \(P_{\text{g}}\) is the covariate-scaled population parameter, and \(\eta_{i}\) is the individual value of IIV for subject \(i\), with \(\eta\) following a normal distribution \(N\left( {0,\omega^{2} } \right)\). Off-diagonal elements of the variance–covariance matrix were evaluated to identify covariances between the individual random effects. Subsequently, correlations between the random effects were derived from the covariances. To account for the difference between observed MCLA-128 concentrations and model-predicted concentrations, a proportional and a combined proportional and additive residual error model were evaluated. Data points below the LLOQ were imputed as LLOQ/2 (0.025 mg/L) and additive residual variability was fixed to this value [12].
2.3.3 Covariate Analysis
Continuous (age, height, body weight, tumor burden, body surface area [BSA] and fat-free mass [FFM] at baseline) and categorical (sex and HER2 status) covariates were evaluated for inclusion in the model. For continuous covariates, missing values were imputed by the median value. Body weight, BSA, and FFM were independently tested for their effect on CL, V1, and Vmax. BSA and FFM were calculated using the following equations [13]:
where WT is body weight in kilograms, HT is height in centimeters, and BMI is body mass index.
Additionally, the magnitude of target expression is expected to affect the capacity of target-mediated clearance. Therefore, HER2 status, determined by fluorescence in situ hybridization, was evaluated as a covariate on Vmax in addition to tumor burden at baseline, defined as the sum of the longest diameters of the target lesions. Age and sex were evaluated on CL and V1. Because body size measures are influenced by sex, sex was only evaluated with body size covariates already included in the model. Continuous covariates were implemented using the following equation:
where \(P_{\text{g}}\) is the covariate-scaled population parameter, \(P_{\text{pop}}\) is the population parameter, \(\theta_{\text{cov}}\) is the covariate effect parameter, and COV is the continuous covariate value. Dichotomous covariates were implemented as follows:
where \(\theta_{\text{cov}}\) represents the fractional change in the population parameter and \({\text{COV}}_{\text{D}}\) is the dichotomous covariate. HER2 status was missing for part of the population (33%). The effect of HER2 status on Vmax was therefore evaluated in two analyses: (1) an analysis including the subpopulation for whom HER2 status was available (n = 83) and (2) estimating a separate Vmax parameter for the missing group. The selected covariates were evaluated using a forward-inclusion and backward-elimination method. For forward inclusion, a significance level of p < 0.01 was used, corresponding to a decrease of objective function value (OFV) of > 6.63. A significance level of p < 0.005 was set for backward elimination, corresponding to an increase of OFV of > 7.88.
2.3.4 Model Evaluations
Models were evaluated based on general goodness-of-fit plots, mechanistic plausibility, stability and precision of parameter estimates, and change in OFV where p < 0.01 was considered significant (OFV drop of > 6.63, with 1 degree of freedom). In addition, visual predictive checks (VPCs) were performed to evaluate the performance of the model. Parameter uncertainty was obtained from the default covariance step in NONMEM and from the sampling importance resampling method [14].
2.3.5 Software
Non-linear mixed-effects modeling was performed using NONMEM (version 7.3.0, ICON Development Solutions, Ellicott City, MD, USA) and Perl-speaks-NONMEM (version 4.4.8) [15, 16]. All models were estimated using the first-order conditional estimation method with η–ε interaction. Pirana (version 2.9.2) was used as graphical user interface [17]. Data handling, graphical evaluation, and simulations were performed using R (version 3.3.1) [18].
2.4 Simulations
To evaluate whether flat dosing of MCLA-128 is appropriate for this patient population, simulations were performed. First, the combinations of body weight, height, sex, and tumor burden for the simulation dataset were randomly sampled (with replacement) from the original dataset (n = 116), with even proportions of male and female subjects. The combinations of weight, height, and sex values were sampled as fixed combinations, to preclude sampling of irrational weight, height, and sex combinations. Tumor burden was not correlated with tumor type or sex. Subsequently, concentration–time profiles of MCLA-128 were simulated for patients receiving a 750-mg flat dose, 420 mg/m2, 11 mg/kg, and 17 mg/kg of FFM MCLA-128 administered once q3wk in an intravenous infusion of 2 h, using the final model. The weight-based dosages were rounded and chosen such that the median BSA, weight, and FFM in the study population (1.78 m2, 68.2 kg and 43.1 kg FFM) corresponded to a flat dose of 750 mg. Each simulation dataset consisted of 3000 patients (n = 1000 per dose group). The area under the concentration-time curve for a dosing interval (AUC0–τ), maximum concentration (Cmax), average concentration (Cave), and trough concentrations (Ctrough) on day 21 were assessed and compared between the flat, BSA-based, and weight-based dosing groups. Simulated negative concentrations were fixed to 0.001 mg/L.
Additional simulations were performed to evaluate the translational and predictive performance of the previously developed preclinical pharmacokinetic model, which was based on data from cynomolgus monkeys and for this purpose allometrically scaled to humans [10]. A VPC was performed using the original clinical dataset including only observations and dose records of patients who received the 750-mg flat dose of MCLA-128. The mean and range of the simulated exposures (n = 100 per patient), defined by AUC0–last timepoint was compared to the observed AUC0–last timepoint, using a non-compartmental analysis.
3 Results
3.1 Data
Pharmacokinetic data were available for 116 patients. Of these 116 patients, 93 patients received a dose of 750 mg q3wk. The remaining patients received 40 mg (n = 1), 80 mg (n = 2), 160 mg (n = 1), 240, 360, and 480 mg (each n = 3), 600 mg (n = 7), and 900 mg (n = 3) of MCLA-128. In total, 1115 observations, with a median [range] of 10 [1–10] observations per patient, were included in the analysis. Overall, 22 of the 1115 observations (2%) were below the LLOQ and fixed to a value of 0.025 mg/L (LLOQ/2). Patient demographics are depicted in Table 1. Height was missing for one female patient with a relative high weight and one male patient with a relative low weight; therefore, the median height of the population (164 cm) was imputed, instead of median height per sex group.
3.2 Pharmacokinetic Model
A two-compartment model with parallel linear and non-linear clearances from the central compartment best described the data (adding a second compartment to a one-compartment model with linear clearance decreased OFV by 4059 points and adding non-linear clearance to a two-compartment model with linear clearance decreased the OFV by 384 points). The non-linear clearance was described using Michaelis–Menten kinetics. The final model structure was defined by the following differential equations:
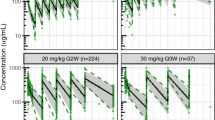
where CL represents the linear clearance, Q is the intercompartmental clearance, V1 is the volume of distribution in the central compartment, V2 is the volume of distribution in the peripheral compartment, A1 is the amount of drug in the central compartment, Vmax is the maximum elimination rate, C1 is the drug concentration in the central compartment, Km is the drug concentration at which half the drug targets are occupied, and A2 is the amount in the peripheral compartment. A combined proportional and additive residual error model described residual variability best. Body size parameters of body weight, BSA, and FFM were all, univariately, identified as significant covariates affecting CL and V1, where FFM provided the best model fit and led to an OFV drop of 131. Fat-free mass explained 8.4% of IIV in CL and 5.6% of IIV in V1. After implementation of sex-specific FFM in the model, sex had no significant additional impact on the pharmacokinetic parameters of MCLA-128. Observed sex differences in concentration–time curves were thus explained by the sex-specific differences in FFM (median of 58.7 kg FFM in men vs 39.8 kg FFM in women). In addition, age and HER2 status had no significant effect on the pharmacokinetics of MCLA-128. Tumor burden was identified to significantly affect the non-linear clearance capacity, explaining 4.7% of IIV in Vmax (OFV drop of 10 points). The parameter estimates of the final model are depicted in Table 2. The 95% confidence intervals of the sampling importance resampling indicated that parameter estimates were precise. In addition, the VPC, stratified on the different dose groups demonstrated a good model fit across the dose range evaluated (Fig. 1).

Visual predictive check (VPC) for MCLA-128 concentrations plotted on a log-scale, stratified on dose. Open circles are observed MCLA-128 concentrations in the first cycle of patients receiving MCLA-128 every 3 weeks, the solid line represents the median of the observed data, the dashed lines represent the 5th and 95th percentiles of the observed data, the shaded areas represent the 95% confidence interval of the simulated median (dark blue) and 5th and 95th percentile (light blue) concentrations (n = 500). For dose groups including three or fewer patients, only the observed concentrations and the 95% confidence interval of the simulated median are depicted
3.3 Simulations
Simulations of the final model with flat, body weight-based, BSA-based, and FFM-based doses demonstrated comparable values of AUC0−τ, Cmax, Cave, and Ctrough concentrations and comparable coefficients of variation (Table 3). In addition, the different dosing strategies demonstrate comparable Ctrough concentrations over different weight groups (Fig. 2), indicating that weight, FFM- or BSA-based dosing does not substantially lower variability in exposure between patients, compared to flat dosing of MCLA-128. Moreover, the effect of body size parameters on CL and V1 on exposure parameters is minimal. Approximately 3% of the simulated concentrations were below zero and fixed to 0.001 mg/L.

Boxplots depict simulated MCLA-128 trough concentrations (Ctrough) for different weight groups and dosing strategies: flat dosing (750 mg), body surface area (BSA)-based dosing (420 mg/m2), body weight (WT)-based dosing (11 mg/kg), and fat-free mass (FFM)-based dosing (17 mg/kg) with n = 1000 per dosing strategy
In addition, to obtain some insight into the expected pharmacological activity after a flat dose of 750 mg given q3wk, target receptor occupancies (RO) were predicted for the mean serum peak and trough concentrations of MCLA-128 (222.7 and 1.40 mg/L, respectively) based on the dissociation constants for HER2 (0.467 mg/L) and HER3 (0.292 mg/L). [2] This resulted in predicted RO percentages of 99.8% and 99.9% at Cmax and 75% and 82.8% for HER2 and HER3 at Ctrough, respectively.
The simulations performed with the preclinical pharmacokinetic model, scaled from cynomolgus monkeys to humans, were compared to the observed pharmacokinetic data in a VPC (Fig. 3, left panel). Observations were slightly overpredicted using the preclinical model. In addition, high variability was observed at the lower concentrations. In the clinical pharmacokinetic model, IIV on Vmax was included, to account for the differences in HER2 or HER3 expression between patients. Including IIV on Vmax in the preclinical model showed a better prediction of the lower concentrations observed in patients (Fig. 3, right panel). In addition, the observed AUC0–last timepoint was in the range of the simulated AUC0–last timepoint (n = 100) for 92% of patients.

Visual predictive check for MCLA-128 concentrations over time. The shaded blue area is the 95% prediction interval of simulated concentrations (n = 1000) using the preclinical pharmacokinetic model (left panel) and the preclinical pharmacokinetic model with inter-individual variability (IIV) on the maximum elimination rate of the non-linear clearance (Vmax) (right panel), patients received a dose regimen of a MCLA-128 750-mg flat dose administered every 3 weeks; open circles are observed MCLA-128 concentrations in the first cycle of patients participating in the 750-mg cohort of the clinical trial
4 Discussion
The pharmacokinetics of MCLA-128 were best described by a two-compartment model with parallel linear and non-linear clearance from the central compartment. The estimated parameters of the final model were in accordance with the general pharmacokinetic characteristics of therapeutic monoclonal antibodies as previously described [11, 19]. The following median (range) values were reported for monoclonal antibodies: 3.1 L (2.4–5.5) and 2.8 L (1.3–6.8) for V1 and V2, respectively, and 0.013 L/h (0.003–0.223) for CL. Estimations for Vmax and Km varied widely among different monoclonal antibodies, with Vmax values ranging from 0.004 to 4.38 mg/h and Km values between 0.033 and 74 mg/L [11]. The Michaelis–Menten estimates for MCLA-128 were within this wide range. The estimate of Km (0.211 mg/L) is also comparable to the dissociation constants for HER2 and HER3, 0.467 mg/L and 0.292 mg/L, respectively [2]. These findings confirm the plausibility of the parameter estimates.
Tumor burden was identified as a significant covariate affecting Vmax. Similar results have been identified previously for trastuzumab [20]. HER2 status, sex, and age were not identified to significantly impact the disposition of MCLA-128. HER3 receptor expression data were not available, possibly explaining why HER2 status alone was not identified as a covariate affecting non-linear clearance capacity. However, HER3 expression is often low [1]. The maximum non-linear clearance capacity depends on the amount of target that is expressed, depending on both HER2 status and tumor size. Because tumor burden is a more variable covariate than HER2 status, tumor burden could be a better determinant of the maximum non-linear clearance capacity, possibly explaining why tumor burden significantly affected Vmax and HER2 expression did not.
Body size measurements (body weight, BSA, and FFM) affected linear CL and V1, where inclusion of FFM led to the best model fit. From a physiological point of view, FFM is also expected to be more closely related to the distribution and linear elimination of monoclonal antibodies than total body weight. Distribution of monoclonal antibodies is mainly limited to blood plasma and extracellular fluids, owing to their size and hydrophilic character. It has been demonstrated that blood volume correlates better with FFM or lean body weight than with total body weight because blood volume does not proportionally increase with body weight [21]. Monoclonal antibodies are metabolized via proteolytic catabolism and intracellular degradation after binding to the target. Proteolytic catabolism is mediated via the FcRn receptor and is a linear process at therapeutic concentrations of monoclonal antibodies [19, 22]. Proteolytic catabolism of immunoglobulin G antibodies takes place in the skin, muscle, liver, and gut tissue [19] and is thus expected to be more closely related to FFM than measures relying on total body weight and size. The estimated effect of FFM on CL and V1 was relatively high, with exponents of 1.2 and 0.8, respectively. However, simulations demonstrated that exposure measures (AUC0–τ, Cmax, Cave, and Ctrough) and variability in exposure measures were comparable between flat dosing and body size-based dosing strategies. Therefore, there appears no rationale for body size-based dosing over flat dosing of MCLA-128 from an exposure perspective, which is in agreement with the findings for other therapeutic monoclonal antibodies in oncology. [23] The predictions for expected HER2 and HER3 RO after a fixed dose of 750 mg of MCLA-128 indicate that full RO is expected at Cmax and that relevant pharmacological activity can still be assumed at the end of a 3-week dosing interval (75% RO for HER2 and 82.8% for HER3).
The observed concentration–time data of the patients in the clinical trial were used to evaluate the predictive value of the preclinical model [10]. The preclinical model slightly overpredicted the observed concentrations of MCLA-128 in patients receiving 750 mg q3wk. Disposition of monoclonal antibodies is affected by the amount of target expression via target-mediated drug disposition. The preclinical model was based on data from healthy cynomolgus monkeys that expressed endogenous HER2 receptors. Tumor-bearing patients are expected to express more HER2 or HER3 receptors, indicating greater and more variable non-linear clearance capacity, potentially leading to lower concentrations of MCLA-128 in humans. Though the estimate of Vmax in the preclinical model was higher compared with the clinical model (0.500 mg/h vs 0.114 mg/h), the variability in the clinical Vmax was much higher. Including this variability in the preclinical model demonstrated that the overprediction of observed MCLA-128 concentrations can be attributed to variability in target expression between patients included in the trial.
To extrapolate the preclinical model, the preclinical CL, Q, Vmax, V1, and V2 parameters were allometrically scaled using a fixed exponent value of 0.75 for CL, Q and Vmax and a value of 1 for V1 and V2. These exponents were fixed because the weight range of the cynomolgus monkeys was narrow and did not allow for appropriate estimation of the exponent value, this might also have contributed to the slight discrepancy between the predicted MCLA-128 concentrations from preclinical data and the observed concentration in the clinical trial. However, this post hoc evaluation of the preclinical model showed that very reasonable and useful estimates of human exposure can be obtained from cynomolgus monkey data to support initial dose selection for these types of monoclonal antibodies.
Data on the generation of anti-drug antibodies were not available at the time of analysis and, therefore, not accounted for in the analysis. However, it can be expected that excluding the formation of anti-drug antibodies from the model did not affect our results, as only pharmacokinetic data of the first treatment cycle were included in the analysis.
5 Conclusions
A pharmacokinetic model was developed that adequately described the pharmacokinetic characteristics of MCLA-128 over a range of doses. Fat-free mass was found to significantly affect CL and V1 and explained part of the IIV in these parameters. However, simulations demonstrated that the impact of body size parameters on the disposition of MCLA-128 was minimal and that flat dosing of 750 mg of MCLA-128 q3wk is expected to provide relevant pharmacological activity in patients with solid tumors. It contributes to the existing evidence that flat dosing is to be preferred for anticancer monoclonal antibodies.
References
Geuijen C, Rovers E, Nijhuis R, Visser T. Preclinical activity of MCLA-128, an ADCC enhanced bispecific IgG1 antibody targeting the HER2:HER3 heterodimer. J Clin Oncol. 2014;32(Suppl.):abstract 560.
Geuijen CAW, De Nardis C, Maussang D, Rovers E, Gallenne T, Hendriks LJA, et al. Unbiased combinatorial screening identifies a bispecific IgG1 that potently inhibits HER3 signaling via HER2-guided ligand blockade. Cancer Cell. 2018;33:922–35.
Arteaga C, Sliwkowski M, Osborne C, Perez E, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011;9:16–32.
Baselga J, Cortes J, Kim S, Im S, Hegg R. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–19.
Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer resistance to trastuzumab in breast cancer. Clin Cancer Res. 2009;15:7479–91.
Wong AL, Lee SC. Mechanisms of resistance to trastuzumab and novel therapeutic strategies in HER2-positive breast cancer. Int J Breast Cancer. 2012;2012:415170.
Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9.
Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–41.
Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, Chakrabarty A, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci USA. 2011;108:5021–6.
de Vries Schultink AHM, Doornbos RP, Bakker ABH, Bol K, Throsby M, Geuijen C, et al. Translational PK-PD modeling analysis of MCLA-128, a HER2/HER3 bispecific monoclonal antibody, to predict clinical efficacious exposure and dose. Invest New Drugs. 2018;36:1006–15.
Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:633–59.
Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481–504.
Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44:1051–65.
Dosne AG, Bergstrand M, Harling K, Karlsson MO. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J Pharmacokinet Pharmacodyn. 2016;43:583–96.
Beal S, Sheiner L, Boeckmann A, Bauer R. NONMEM 7.3.0 users guides. Hanover (MD): ICON Development Solutions. 2013.
Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN): a Perl module for NONMEM related programming. Comput Methods Progr Biomed. 2004;75:85–94.
Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometr Syst Pharmacol. 2013;2:e50.
R Core Team 2018. The R Project for statistical computing. Vienna, Austria. https://www.r-project.org/. Accessed 11 Jan 2020.
Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493–507.
Bernadou G, Campone M, Merlin JL, Gouilleux-Gruart V, Bachelot T, Lokiec F, et al. Influence of tumour burden on trastuzumab pharmacokinetics in HER2 positive non-metastatic breast cancer. Br J Clin Pharmacol. 2016;81:941–8.
Boer P. Estimated lean body mass as an index for normalization of body fluid volumes in humans. Am J Physiol. 1984;247:F632–6.
Mould D, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies. Biodrugs. 2010;24:23–39.
Hendrikx JJMA, Haanen JBAG, Voest EE, Schellens JHM, Huitema ADR, Beijnen JH. Fixed dosing of monoclonal antibodies in oncology. Oncologist. 2017;22:1212–21.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Kees Bol, Robert P. Doornbos, Anastasia Murat, and Ernesto Wasserman are employees of Merus N.V. Aurelia H.M. de Vries Schultink, Thomas P.C. Dorlo, Jan H.M. Schellens, Jos H. Beijnen, and Alwin D.R. Huitema have no conflicts of interest that are directly relevant to the content of this article.
Consent to participate
All patients provided written informed consent before study entry.
Funding
This research was funded by Merus N.V. Thomas P.C. Dorlo is personally supported by NWO/ZonMw through a Veni grant (grant no. 91617140).
Ethics approval
This study was conducted in compliance with the Declaration of Helsinki, International Conference on Harmonization Guidelines for Good Clinical Practice. The protocol was approved by the ethics committees of all participating centers.
Rights and permissions
About this article

Cite this article
de Vries Schultink, A.H.M., Bol, K., Doornbos, R.P. et al. Population Pharmacokinetics of MCLA-128, a HER2/HER3 Bispecific Monoclonal Antibody, in Patients with Solid Tumors. Clin Pharmacokinet 59, 875–884 (2020). https://doi.org/10.1007/s40262-020-00858-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00858-2