
Abstract
Immune checkpoint inhibitors are a new class of monoclonal antibodies that amplify T-cell-mediated immune responses against cancer cells. The introduction of these new drugs, first anti-cytotoxic T-lymphocyte-associated protein 4 (anti-CTLA4) and then anti-programmed death-1 (anti-PD1), was a major improvement in the treatment of advanced or metastatic melanoma, a highly immunogenic tumour. The development strategy for immune checkpoint immunotherapies differed from that traditionally used for cytotoxic therapies in oncology. The choices of doses at which to conduct clinical trials, and subsequently the choice of doses at which to use these new therapies, were not based on the identification of a maximum tolerated dose from dose-escalation studies; thus, pharmacokinetic and pharmacokinetic–pharmacodynamic modelling was essential. The studies conducted have shown that the pharmacokinetics of ipilimumab were linear and not time-dependent. In addition, there was a correlation between the trough concentrations of ipilimumab and its therapeutic efficacy. On the contrary, the anti-PD1 immunotherapies nivolumab and pembrolizumab had time-dependent pharmacokinetics. Their therapeutic efficacy was not related to their trough concentration, but there was a correlation between the clearance of anti-PD1 and the survival of melanoma patients. This review highlights the complexity of interpreting the exposure–response relationships of these agents. Further studies are needed to assess the value of therapeutic drug monitoring of immune checkpoint inhibitors in the treatment of melanoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The incidence of skin melanoma is growing faster than that of any other solid tumour—160,000 new cases and 48,000 deaths occur each year worldwide [1]. Melanoma develops a complex interaction with the immune system, highlighted by several observations: melanoma antigens are recognized by T cells [2]; immune infiltrations in primary tumours have a strong prognostic importance [3]; spontaneous regression is sometimes observed in metastatic patients [4]; and vitiligo may be associated with regression of metastatic lesions under treatment [5, 6].
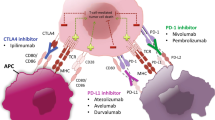
Immune checkpoint inhibitors are a new class of immunotherapies whose discovery was recently (2018) awarded a Nobel Prize in medicine. Drugs targeting the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or programmed death-1 (PD-1) receptor represent a major advance in the treatment of advanced or metastatic melanoma. Indeed, to prevent autoimmunity, several signalling pathway control points regulate T-cell activity at different steps of the immune response in a process called peripheral tolerance. The CTLA-4 and PD-1 control points are at the heart of this process; CTLA-4 is currently considered the leader of immune checkpoint inhibition because it intercepts self-reactive cells at the initial step of T-cell activation, typically in the lymph nodes [7, 8], and the PD-1 receptor pathway regulates already active T cells at later steps of the immune response process, primarily in peripheral tissues [7]. In fact, PD-1 expression is the characteristic sign of ‘exhausted’ T cells that have experienced significant levels of stimulation or reduced CD4+ T-cell aid [9]. A high level of PD-1 expression is found in nonfunctional T cells in the context of chronic infection [10] or tumour progression [11].
The distribution of PD-1 ligands differs from those of CTLA-4, which has direct consequences on the efficacy and tolerability profile of the various immune checkpoint inhibitor monoclonal antibodies used in oncology. Unlike anti-CTLA-4, PD-1/PD-L1 (programmed death-ligand 1) interaction occurs more selectively at the level of the tumour microenvironment, regulating the effective phase of the T cell response, making it a more attractive control point to target. In humans, the small lymphocyte subpopulation affected by PD-1 blockade, compared with that affected by CTLA-4 blockade, may explain the lower incidence of adverse events (AEs) seen to date with anti-PD1 versus anti-CTLA-4 [12].
The indications for immune checkpoint inhibitors authorized in the treatment of melanoma differ according to their target antigen. Anti-PD1 immunotherapy is recommended as the first-line treatment for advanced nonresectable or metastatic melanoma, regardless of BRAF V600 status, and is indicated as second-line treatment for BRAF V600-mutated patients after failure of targeted anti-BRAF and anti-MEK combination therapies [13, 14]. Anti-CTLA4 immunotherapy is indicated in the first line in combination with anti-PD1, and as monotherapy in the second line of treatment after failure of anti-PD1 treatment. Anti-PD1 and anti-CTLA4 are also indicated in the adjuvant treatment of melanoma after surgical resection, although, in view of its toxicity, the use of an anti-CTLA4 in adjuvant therapy is controversial [15].
The purpose of this review is to present and analyse currently available pharmacokinetic (PK) and pharmacodynamic (PD) data of new immune checkpoint inhibitors developed in skin melanoma, and to identify the doses and schedules of administration that optimize the best efficacy/tolerance ratio.
2 Structure of Monoclonal Antibodies
2.1 Approved Monoclonal Antibodies
Immune checkpoint inhibitors currently approved in Europe and the US for the treatment of advanced or metastatic melanoma are ipilimumab (Yervoy®, Bristol-Myers Squibb) [16], an anti CTLA-4, and pembrolizumab (Keytruda®, Merck) [17] and nivolumab (Opdivo®, Bristol-Myers Squibb) [18], both anti-PD-1 agents (Table 1). Other anti-PD-1 agents, such as cemiplimab (Libtayo®, Sanofi-Regeneron) currently approved in the US for the treatment of patients with metastatic cutaneous squamous cell carcinoma or locally advanced cutaneous squamous cell carcinoma who are not candidates for curative surgery or curative radiation [19], will follow.
Ipilimumab is a fully human immunoglobulin (Ig) G1 kappa monoclonal antibody directed against the CTLA-4 protein [20]; pembrolizumab is a humanized IgG4 monoclonal antibody directed against the PD-1 receptor [21, 22]; and nivolumab is a fully human IgG4 monoclonal antibody also directed against the PD-1 receptor [23, 24].
2.2 Other Molecules
A few small molecule PD-1/PD-L1 inhibitors have also been described to date, i.e. peptidic direct PD-1 antagonists, small molecule PD-L1 dimerizers, and small molecules whose mode of action remains unknown [25], but most of them have only modest inhibitory activity [26].
Monoclonal antibodies are very specific to their targets and provide a long-lasting effect, but they also have certain disadvantages in the context of immunotherapy. Their large size makes it difficult to access exhausted intratumoural T cells (at least for anti-PD1), and the use of antibodies can be challenging to treat tumours located at immune-privileged sites such as the eye or the brain [27, 28]. In addition, the intravenous route of administration requires patients to go to a hospital. An increasing number of monoclonal antibodies are administered subcutaneously, providing a better quality of life for patients and reducing the economic burden of treatment [29]. These limitations provide arguments for the development of nonmonoclonal antibody immune checkpoint inhibitors, but these drug candidates are outside the scope of this review.
3 Pharmacokinetics
3.1 Ipilimumab
The PK of ipilimumab have not been evaluated in healthy volunteers, with the majority of available data coming from patients with advanced melanoma.
The PK of ipilimumab were studied by population approach (popPK) following a phase II randomized controlled dose-escalation clinical trial (CA184-022) (Table 2) [30]. This analysis provided the information submitted to the European Medicines Agency (EMA) for the approval of ipilimumab [20, 31].
Modelling results showed linear, nontime-dependent (time-invariant) PK for the dose range considered (0.3–10 mg/kg). Plasma ipilimumab concentration–time data were satisfactorily described by a bicompartmental model, with zero-order intravenous infusion and first-order elimination. After intravenous administration, ipilimumab followed biphasic elimination, with a distribution half-life of 27.4 h, and a slow elimination, with an average half-life of 14.7 days. The clearance averaged 0.36 L/day (normalized to a body weight of 80 kg). Volume of distribution of the peripheral (Vp) and central compartments (Vc) was estimated at 4.15 and 3.11 L, respectively (normalized to a body weight of 80 kg).
Body weight at baseline was the most influential covariate on clearance and Vc, which was consistent with the nonspecific monoclonal antibody removal mechanism mediated by the reticuloendothelial system [32, 33]. The effects of other covariates were within ± 20%, which led the authors to consider them as not clinically significant. A 22% increase in clearance was estimated due to anti-ipilimumab antibodies [anti-drug antibody (ADA)], but was not considered as clinically significant by the authors as < 5% of patients had developed ADAs, most of which were transient. Only body weight at baseline and lactate dehydrogenase (LDH) concentration were retained as covariates in the final model.
3.2 Pembrolizumab
The PK of pembrolizumab were first described using a time-invariant model that provided the information found in the Keytruda monograph. The modelling proposed by Ahamadi et al. in 2017 with pembrolizumab was performed from data obtained at doses ranging from 1 to 10 mg/kg administered intravenously [34]. Most patients were treated for melanoma or nonsmall cell lung cancer (NSCLC) but several other tumour types were found in the KEYNOTE-001 first-in-human dose-escalation study (Table 2).
The PK of pembrolizumab were well-described using a bicompartmental model with linear clearance. Nonlinear PK were observed for pembrolizumab at doses well below 1 mg/kg [35]. However, a trend for increasing concentrations beyond week 20 was detected, which may indicate a contribution of time to the value of PK parameters. The estimated elimination half-life was 27.3 days. The clearance was low (0.22 L/day) and the volume of distribution was estimated at approximately 6 L, a result compatible with limited distribution outside the extracellular space. The effects of albumin at baseline, sex, and history of ipilimumab treatment on Vc were retained in the final model. The effects of albumin and tumour load at baseline, as well as glomerular filtration rate, sex, tumour type, Eastern Cooperative Oncology Group performance status (ECOG-PS) score at baseline, and ipilimumab treatment history on clearance were also retained in the final model. The ECOG-PS score is the standard criterion for measuring how the disease impacts a patient’s daily living abilities in terms of their ability to care for themself, as well as daily activity, and physical ability. Grade 0 translates to a fully active patient, able to carry on all predisease performance without restriction, while grade 5 translates to death.
The time-dependent PK hypothesis led to a new popPK analysis (Table 2) [36]. Several covariates were retained in the time-dependent model. Pembrolizumab clearance was associated with low albumin levels, greater tumour size at baseline and a higher ECOG-PS score. Systemic inflammation, cachexia and a target-mediated drug disposition (TMDD) component of elimination were among the hypotheses put forward by the authors to explain these observations.
The median population clearance value was approximately 20% lower at steady state compared with clearance at first administration. According to the authors, this decrease of clearance should not have clinical consequences because variations in exposure by a factor of 5 were observed in clinical trials without any consequence on the efficacy or safety of pembrolizumab.
3.3 Nivolumab
The PK of nivolumab were studied using a popPK approach using data from 1895 patients from three phase I studies, three phase II studies, and five phase III studies (Table 2) [37]. Most patients were treated for melanoma or NSCLC, but several other tumour types were also found in these studies.
Patients received nivolumab at doses ranging from 0.1 to 10 mg/kg in single administrations every 2 or 3 weeks depending on the study. The majority of patients received nivolumab 3 mg/kg every 2 weeks.
The PK of nivolumab were described as linear, with a dose-independent clearance from 0.1 to 20 mg/kg. The authors did not present any external validation of their model. The inclusion of a TMDD component in the elimination did not improve the model. The selected model was a bicompartmental model with zero-order intravenous administration and first-order elimination. The PK of nivolumab were initially modelled with invariant clearance, but were later reassessed based on findings with pembrolizumab.
In the final model, clearance decreased over time, with a maximum change from baseline of approximately 24.5%. The time-dependent component of nivolumab clearance was described by a sigmoid relationship. Based on the results of this analysis, nivolumab followed biphasic elimination after intravenous administration consisting of a rapid distribution phase, with a half-life of 32.5 h, and a slow elimination phase, with an average half-life of 25 days at steady state. The average clearance was estimated at 0.226 L/day (normalized to a body weight of 80 kg), and the Vc and Vp were estimated at 3.63 and 2.78 L, respectively (normalized to a body weight of 80 kg, White female).
The US FDA analysis revealed that the clearance of nivolumab decreases as the disease status improves [38]; the change in clearance of nivolumab is correlated with the post-treatment ECOG-PS score and is not just time-dependent. By studying patient data from the study by Bajaj et al. [37], it was shown that patients with better status and higher survival had a greater reduction in nivolumab clearance compared with their clearance at baseline. This reduction resulted in significantly greater steady-state exposure than with the first administration of nivolumab.
4 Pharmacodynamics
4.1 Ipilimumab
A phase II randomized controlled exploratory study that studied the impact of ipilimumab on the tumour microenvironment showed an increase in T-cell activation marker expression [39]. The mean levels of activated (HLA-DR+), CD4 and CD8 peripheral blood T cells were increased after ipilimumab administration. No differences in activation were observed between the 3 and 10 mg/kg doses. These results were consistent with a dose-escalation study conducted in 46 patients with metastatic melanoma (stage IV) [40].
The absolute lymphocyte count, a measure of all circulating B and T lymphocytes, was positively associated with overall survival (OS) in melanoma, in therapeutic trials with ipilimumab [41]. Several studies also showed an increase in the absolute number of circulating lymphocytes qualified as a PD marker of immune cell activation by ipilimumab at doses of 3 and 10 mg/kg [30, 39, 41]. The absolute number of lymphocytes increased in a dose-dependent manner and continued to increase during the induction period.
Study CA184-022 was a randomized controlled, phase II, dose-escalation clinical trial (Table 3). Ipilimumab showed a dose-dependent effect on OS. Favourable, albeit nonsignificant, results were noted in favour of the 10 mg/kg dose, both in terms of response and OS. Similarly, the incidence of immune-related AEs (irAEs) of any grade increased with increasing doses of ipilimumab. No grade 4 toxicity was found and no grade 3–4 toxicity occurred for the 0.3 mg/kg dose. The most common cause of treatment interruption or death was disease progression.
The exposure–response (E–R) relationship of ipilimumab in patients with advanced (unresectable or metastatic) melanoma was evaluated in a retrospective study (Table 3) [42]. The minimum concentration at steady state (Cminss) at the end of the induction phase (four doses spaced 3 weeks apart) was a statistically significant predictor of response and there was a statistically significant relationship between ipilimumab exposure and the hazard ratio of death.
The E–R analysis for tolerability showed that the probability of having a grade 2 or higher irAE, and the probability of a first irAE occurring at any time, increased with Cminss over the studied dose range.
Given the association observed between Cminss and both therapeutic efficacy and tolerability, dose individualization could be an effective approach for ipilimumab treatment in patients with advanced or metastatic melanoma, if a range of optimal concentrations in terms of therapeutic efficacy and tolerability is determined.
4.2 Pembrolizumab
A first dose-escalation clinical trial, involving 13 patients, focused on elucidating the PK/PD relationship by measuring the response in terms of interleukin (IL)-2 release over a range of 0.005–10 mg/kg [43]. The biologically active dose was estimated at 2 mg/kg as the simulation results approached saturation at exposures consistent with this dose [44].
To determine the lowest effective dose to be used in the KEYNOTE-001 phase I clinical trial, a PD study of pembrolizumab was conducted in mice [45]. In order to describe the PK/PD of pembrolizumab, a complex model was developed from experimental mouse data and specific mouse physiological parameters from the literature. For human dose–response simulations, the model was translated by replacing mouse parameters with human parameters where possible, and allometrically modifying mouse parameters or keeping them constant when the human parameters were unknown.
The PK model used in humans was the same as that used in the work of Elassaiss-Schaap et al. [46].
Simulations showed that the probability of reaching a more than 30% reduction in tumour size reached a plateau for doses ≥ 2 mg/kg every 3 weeks. A minor increase in benefit was predicted by administration every 2 weeks compared with every 3 weeks.
Modelling PD-1 occupancy at the tumour level indicated that PD-1 was saturated at clinically relevant concentrations.
KEYNOTE-001 PK/PD data were limited and left uncertainties regarding the linearity of pembrolizumab PK and PD. In order to allow for selection of the lowest dose for future clinical trials, the choice of the design of an additional cohort of KEYNOTE-001 (A2) was guided by modelling and simulation results [35].
PDs were evaluated by measuring the IL-2 stimulation ratio in blood, assuming that the IL-2 stimulation ratio would be a surrogate marker for pembrolizumab binding to the PD-1 target, a reflection of pembrolizumab binding to its target at the tumour level, and, ultimately, a marker of pembrolizumab antitumour efficacy. The potency of pembrolizumab (half maximal inhibitory concentration) as measured using the IL-2 stimulation ratio test was 0.54 mg/L (95% confidence interval (CI) 0.12–2.3 mg/L).
The results of the simulations conducted with the consolidated PK/PD model (after the introduction of the A2 cohort data) showed that target engagement increased monotonously. A dose of 2 mg/kg every 3 weeks was required to achieve 90% probability of 95% engagement at steady state [35].
To characterize the kinetics of tumour size change during treatment, and to identify sources of variability in response to pembrolizumab, a tumour growth model of melanoma was developed (Table 3) [47].
The areas under the curve (AUCs) for the pembrolizumab concentration–time curves were obtained from the popPK results [34]. The AUC over 6 weeks at steady state (AUCss6w) was selected as the metric, to account for interdose duration differences.
The initial model (based on the KEYNOTE-001 study) had to take into account the marked heterogeneity of responses. Patients who responded typically showed an early (slow or rapid) decrease in tumour size, while patients who progressed tended to do so rapidly and to discontinue treatment early. Tumour growth or regression parameters were estimated in a manner comparable with that developed by Claret et al. in their model [48, 49], modified to account for the many patients in whom tumour size remained stable for long periods after an initial decrease, a different pattern from that observed with conventional chemotherapy, where relapse is more classic.
The effect of PD-L1 expression and baseline tumour size on the tumour regression rate, the effect of ipilimumab treatment history and tumour size on the proportion of target tumour tissue available for treatment, and the effect of BRAF mutation status on the tumour growth rate explained some of the interindividual variability of these parameters. There was overlap in the estimates of these parameters between the different groups, with the selected covariates not being predictive of response for an individual patient. The authors indicated that these results suggested that all patients, regardless of their BRAF, PD-1 or ipilimumab treatment history, were likely to benefit from pembrolizumab treatment. Exposure characterized by AUCss6w was not a significant predictor of tumour regression. The simulations showed a relatively flat E–R relationship close to the maximum efficacy plateau. Simulations at 1 mg/kg every 3 weeks suggested that patients with a 50% reduction in pembrolizumab exposure would maintain therapeutic efficacy. However, the correlation of the dynamics of tumour size evolution to survival still needs to be demonstrated.
Overall, these results suggested that there was no significant exposure–effect relationship for pembrolizumab at the doses studied, in favour of an exposure close to the maximum efficacy plateau. In addition, the safety profile was similar for all regimens tested in melanoma clinical trials [50, 51], and a flat E–R relationship was identified for all these regimens in the assessment of irAEs [52].
Nevertheless, the time-dependent PK identified in the work of Li et al. [36] led to questions about the validity of the results of E–R analyses at steady state. Li et al. hypothesized that variations in clearance with time could be seen as a sign that effective treatment was reducing the severity of the disease. Additional evidence to support this assertion was provided by the association between the best overall response (BOR) category of pembrolizumab and the estimated maximum clearance change. Decreasing CL during treatment was associated with better outcomes. The association between clearance variation and BOR also led to an association between clearance variation and OS. Recently, an E–R analysis studied the exposure–survival relationship of pembrolizumab for the first time [53], using data from the KEYNOTE-002 and KEYNOTE-010 studies (Table 3). The findings of the analysis were confirmed prospectively with the results of the KEYNOTE-024 study. To avoid the pitfalls of the correlation between the clearance and efficacy of pembrolizumab, the exposure measure studied was the 6-week standardized first-dose AUC. Clearance estimates were derived from the time-dependent PK model [36]. The results of this analysis showed that for both dose levels, the median survival of patients with the lowest first-dose clearance (first quartile) was more than doubled compared with patients with the highest first-dose clearance (fourth quartile). There would be no causal relationship between exposure and survival, and, in this context, the clearance of anti-PD1 could be an independent marker of disease severity.
4.3 Nivolumab
The cohort extension of a phase Ib, open-label, dose-escalation study (MDX1106-03, NCT00730639) [54] involved 306 patients (Table 3). Efficacy endpoints were objective response rate (ORR) and progression-free survival rate at 24 weeks (PFS24w). ORR was based on the BOR, evaluated according to the Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 criteria.
The dose–response relationship of tolerability was investigated by assessing the correlation between grade 3 or higher AEs, AEs leading to discontinuation of treatment, and exposure to nivolumab.
The dose–response relationship of therapeutic efficacy was evaluated in light of confirmed objective responses and tumour growth dynamics. The E–R relationship between ORR and Cminss was evaluated by separate logistic regression models for each tumour type among melanomas, NSCLC, and renal cell cancer. The tumour growth dynamics were characterized using a previously published nonlinear mixed-effects model [55].
No maximum tolerated doses were identified up to the highest dose studied, i.e. 10 mg/kg every 2 weeks. Overall, nivolumab was considered safe and tolerable up to this dose. The median duration of treatment for all tumour types and doses was 16.1 weeks.
The nature, frequency and severity of AEs were comparable across dose levels and tumour types. The most common cause of treatment discontinuation was disease progression (n = 193, 67.5%). Overall, AEs were manageable and reversible with the introduction of immunosuppressants.
No dose–response relationship was found for AEs at the doses studied. In the 69 melanoma patients evaluated, the PD-1 occupancy rate on peripheral lymphocytes was saturated at doses ≥ 0.3 mg/kg after 8 weeks, but the correlation between peripheral, intratumoural PD-1 binding and cell proliferation was not demonstrated.
The E–R relationship for efficacy was assessed through ORR and tumour growth dynamics (unaffected by unconventional responses observed with onco-immunotherapies). A trend was observed between high Cminss and ORR, but this effect appeared to plateau at 1 mg/kg in melanoma and 3 mg/kg in NSCLC. These results were based on a small number of patients per dose level.
Exploratory analyses revealed that at a given dose level, responder patients were aggregated at the highest levels of the observed Cminss interval. Some patients responded better than patients who received higher doses and had higher nivolumab concentrations.
There was an apparent contradiction between the linear PK of nivolumab, the presence of a correlation between dose and ORR, and the absence of correlation between concentration and ORR or the existence of an E–R correlation but only within a dose range. This result can now be explained by the relationship shown between nivolumab clearance and response to treatment [38].
The authors concluded that nivolumab was well tolerated up to 10 mg/kg every 2 weeks, and E–R relationships for efficacy suggested that nivolumab 1 mg/kg every 2 weeks could be active for highly immunogenic tumours such as melanoma. However, they suggested that a dose of 3 mg/kg every 2 weeks may be necessary for less immunogenic tumours such as NSCLC. Therefore, 3 mg/kg every 2 weeks was used as a single nivolumab monotherapy dose for all tumour types.
The analysis of the E-R relationship in advanced melanoma has since been performed from exposure to nivolumab following first administration (Table 3) [56]. This analysis contributed to the authorization of nivolumab in the treatment of advanced melanoma since the favourable benefit–risk profile of the proposed dosage was supported by this study. A previously published PK model was applied to patient PK data [37]. For exposure–effectiveness analyses, concentrations averaged over time after the first administration (Cavg1) were determined, for each patient, from Bayesian maximum a posteriori estimates of individual PK parameters obtained from the PK model. These were calculated by dividing the AUC after the first dose by the interval between doses (14 days for administration every 2 weeks). The efficacy criterion was the BOR according to the RECIST criteria. Cavg1 was also used as an exposure measure to study the E–R relationship with OS and the time before an AE leading to discontinuation of treatment or death (AE-DC/D).
No correlation was found between Cavg1 and ORR or OS, and none of the covariates studied were significantly correlated with OR. Significant predictors of OS were nivolumab clearance, baseline body weight and baseline LDH. A sensitivity analysis excluding clearance from the full model showed that the effect of Cavg1 on OS remained nonsignificant. Thirty-seven AEs led to treatment discontinuation or patient death among those patients treated for advanced melanoma included in the analysis (one AE leading to death). Cavg1 in nivolumab was not significantly correlated with the risk of AE-DC/D; however, the risk of AE-DC/D was higher for the higher LDH values at baseline. Subsequently, the E–R relationship analysis of Wang et al. [56] was extended to OS in previously untreated severe melanoma patients (Table 3) [57].
No significant effect of Cavg1 on the risk of death was demonstrated. Covariates with a significant effect on OS were ECOG status, baseline body weight, nivolumab clearance, age, and baseline LDH level. The predictor associated with the most important effect was clearance of nivolumab. A sensitivity analysis excluding clearance from the full model found that Cavg1 was not a significant predictor of OS; the effect associated with ECOG status was more important in this analysis, suggesting that the effect of clearance is related to disease severity, as observed for pembrolizumab.
This E–R model was used to propose to the FDA a change in the recommended US dose of nivolumab, from 3 mg/kg every 2 weeks to a fixed dose of 240 mg every 2 weeks.
4.4 Combination of Ipilimumab and Nivolumab
The effects of a combination of nivolumab with ipilimumab in a concomitant or sequential protocol were evaluated in a phase I dose-escalation study (NCT01024231) [12] in advanced or metastatic melanoma. Fifty-three patients received the concomitant protocol and 33 patients were included in the sequential protocol. The results of this trial supported the superiority of the concomitant protocol over the sequential protocol. The safety and response profile at the different dose levels studied in the concomitant protocol contributed to the selection of nivolumab 1 mg/kg in combination with ipilimumab 3 mg/kg.
Following these results, a randomized controlled phase II study (CHECKMATE 069, NCT01927419) involving 142 previously untreated metastatic melanoma patients [58] compared ipilimumab 3 mg/kg with or without nivolumab 1 mg/kg every 3 weeks for four administrations, followed by a maintenance phase with nivolumab 3 mg/kg or placebo every 2 weeks until disease progression or occurrence of intolerable AEs. The primary endpoint was the ORR in patients with nonmutated tumours for BRAF V600. The comparison showed a significantly higher response rate in the arm that combined ipilimumab with nivolumab (61%, 44/72 patients) compared with the arm in which patients were treated with ipilimumab alone (11%, 4/37 patients). The frequency of grade 3 or 4 AEs was also higher—54% in the combination therapy arm versus 24% in the ipilimumab-alone arm.
A phase III randomized controlled trial (CHECKMATE 067, NCT01844505) compared 945 patients with advanced nonresectable or metastatic melanoma not previously treated with nivolumab monotherapy, nivolumab plus ipilimumab, or ipilimumab monotherapy [59]. The median PFS was higher in the dual-therapy group at 11.5 months (95% CI 8.9–16.7) compared with 6.9 months (95% CI 4.3–9.5) in the nivolumab monotherapy arm, and particularly in comparison with ipilimumab monotherapy in which the median PFS was 2.9 months (95% CI 2.8–3.4). As in the study by Postow et al., the frequency of grade 3 and 4 AEs was higher with dual therapy compared with ipilimumab monotherapy, and particularly in comparison with nivolumab monotherapy.
For patients with PD-L1 tumour expression, there was no difference in survival between the nivolumab-alone arm or the nivolumab plus ipilimumab combination; however, for patients without PD-L1 tumour expression, the median PFS was prolonged with dual therapy, at 11.2 months versus 5.3 months.
The results of these two trials (CHECKMATE 067 and 069) led to the extension of the approval of nivolumab for the treatment of advanced melanoma (nonresectable or metastatic) in combination with ipilimumab.
4.5 Combination of Ipilimumab and Pembrolizumab
The results obtained by the combination of ipilimumab and nivolumab motivated the study of the association between ipilimumab and pembrolizumab in advanced melanoma. In an open-label, phase Ib clinical trial (KEYNOTE-029, NCT02089685) [60], 153 patients with advanced melanoma received pembrolizumab 2 mg/kg in combination with ipilimumab 1 mg/kg every 3 weeks for four administrations, followed by a maintenance phase of pembrolizumab 2 mg/kg every 3 weeks for 2 years or until disease progression or intolerable toxicity. The primary endpoints were therapeutic efficacy quantified by ORR, and treatment tolerability. OS and PFS were also collected. Early results from this study showed a response rate of 61% (95% CI 53–69) in patients receiving dual therapy, comparable with that observed with ipilimumab plus nivolumab. Grade 3–4 AEs accounted for 45% of patients. Several randomized, phase II trials studying ipilimumab plus pembrolizumab in combination with different dosages are ongoing.
5 Fixed Dosing
5.1 Pembrolizumab
Based on the PK data for pembrolizumab, Merck sought to re-evaluate the need for dosage adjustment based on patient body weight [52]. The effect of body weight on the PK, described by Ahamadi et al [34], was based on data from 1622 patients with a wide distribution of body weights, with a median body weight of 77.2 kg over a range of 35.7–209.5 kg.
Patient data from clinical trials where pembrolizumab was administered at a dose adjusted to body weight (2–10 mg/kg every 2–3 weeks), and trials where pembrolizumab was administered at the fixed dose of 200 mg every 3 weeks, were used for this analysis. A previously described popPK model [34], based on patient data from the KEYNOTE-001, 002 and 006 studies, was used to estimate the PK parameters and exposures from patient specimens, and to simulate fixed-dose PK. The PK model parameters were re-estimated by incrementing the database with information from ulterior trials.
A relatively flat exposure–effect relationship was observed in the treatment of melanoma and NSCLC in terms of response to tumour size and the occurrence of AEs. The 2 mg/kg every 3 weeks dose was selected for the treatment of melanoma and NSCLC as sufficient to achieve significant clinical benefit, with limited benefit from dose increases. Simulations showed superposition of pembrolizumab exposures for doses of 2 mg/kg every 3 weeks and a 200 mg fixed dose every 3 weeks. PK data from clinical trials conducted at the 200 mg every 3 weeks dose confirmed the results of the simulations.
More recently, the same strategy has been used to obtain EMA approval for the 400 mg every 6 weeks regimen (Table 1). The exposure predicted by the simulations for this new regimen was comparable with that obtained with the 200 mg every 3 weeks and 2 mg/kg every 3 weeks doses [61]. A clinical trial at this dosage regimen to evaluate pembrolizumab as adjuvant therapy in the treatment of advanced cutaneous squamous cell carcinoma is ongoing (KEYNOTE-630).
5.2 Nivolumab
A similar approach was implemented by Bristol-Myers Squibb to identify a fixed dose, unrelated to patient body weight, at which nivolumab should be administered [62]. Body weights from 3458 patients included in 18 clinical trials of nivolumab for various tumours such as melanoma, NSCLC, renal carcinoma, urothelial carcinoma, gastric cancer and small cell bronchial cancer were used to assess the distribution of patient body weights. The fixed dose was chosen to ensure a significant rate of overlap between the fixed dose and the dose-to-weight exposures of nivolumab over this body weight range.
The benefit–risk profile of the fixed dose was evaluated by comparing the exposures obtained with those of the 3 mg/kg every 2 weeks dose, for overall body weight and tumour type distribution, considering the observed safety of use for exposures associated with the 10 mg/kg every 2 weeks dose, the observed safety of use at 3 mg/kg every 2 weeks per body weight group in patients with melanoma, NSCLC and renal cell carcinoma, and the results of the E–R safety and efficacy analyses.
The fixed dose of 240 mg was selected by multiplying the authorized dose of 3 mg/kg every 2 weeks by the median body weight of the population tested, i.e. approximately 80 kg.
This study, based entirely on modelling and simulation, without independent testing in new patients, allowed nivolumab to be administered at a dose of 240 mg every 2 weeks in the treatment of melanoma, NSCLC, renal cancer and urothelial cancer in the US. The exposure, safety and therapeutic efficacy of fixed-dose nivolumab were considered to be similar to those observed with the previously authorized dose per kilogram.
The same strategy based on modelling and simulation of PK data was used to obtain approval for the 480 mg every 4 weeks regimen [63].
6 Discussion
PK and PK/PD studies were at the heart of the development of monoclonal antibody immune checkpoint inhibitors that have now been approved for the treatment of advanced melanoma. The choice of doses at which to conduct clinical trials and, furthermore, the choice of doses at which to use these new therapies were not based on the identification of a maximum tolerated dose from dose-escalation studies. Thus, the strategy for the development of immunotherapies differs from that traditionally applied to cytotoxic therapies in oncology, and PK and PK/PD modelling is essential.
PK studies of anti-PD1 (Table 2) have shown that their clearance varied over time [36,37,38] and that it was correlated with disease progression. The clearance of anti-PD1 decreases as tumour mass decreases. This relationship between clearance and outcome makes the study of exposure–effect relationships complex and the mechanism responsible for this relationship is not yet clear. Cachexia syndrome frequently associated with advanced cancer diseases has been proposed as one of the causes of this phenomenon. In cachexia syndrome, protein catabolism is significantly increased [64], which could potentially have an impact on the degradation of therapeutic IgG anti-PD1. The catabolic state associated with cachexia may decrease with regression of the disease, which would explain the decrease in clearance in patients with reduced tumour mass; however, this hypothesis does not explain the time-independent clearance described for ipilimumab [31]. An important cause of PK variability of monoclonal antibodies is TMDD [65], which is frequently responsible for nonlinear PK. The difference in expression between CTLA4, which is only present on the surface of activated T cells, and PD1, which is expressed on the surface of a wide range of immune cells, has been suggested to explain the time-independent clearance observed with ipilimumab [66].
Another element that made the study of E–R relationships complex was the selection of a relevant endpoint. OS is the criterion that provides the most reliable information, but the median OS is achieved late in patients treated with checkpoint inhibitors. Surrogate endpoints such as PFS or ORR were used to allow early E–R studies to be carried out. It is now known that the correlation for immunotherapies in melanoma treatment between OS and PFS (R2 = 0.192) is poor, as is the correlation between OS and ORR (R2 = 0.028) [67]. The indirect effect of treatment through immune system cells is responsible for atypical responses, i.e. delayed responses, or even initial progression of the tumour mass before the response is obtained. New criteria, such as the immune-related response criteria (irRC), which are better correlated with exposure, have been developed to address the mismatch between PFS/ORR and OS [68]. For pembrolizumab, it has been suggested that the evaluation of response according to the ORR may underestimate the benefit of treatment in approximately 15% of patients [69], while the use of irRC could prevent premature discontinuation of anti-PD1 therapy. However, the correlation between irRC and OS remains to be demonstrated.
Several aspects remain to be studied regarding these new drugs. A recent Danish study [70] found that 55% of the population with advanced melanoma who were eligible for treatment with immune checkpoint inhibitors were not included in clinical trials. An ECOG-PS score ≥ 2, or the presence of brain metastasis, accounted for 74% of the causes of non-eligibility, whereas E–R studies conducted to date have shown a significant impact of the ECOG-PS score on the PK of anti-PD-1. Evaluation of the PK/PD of immunotherapies in patients treated in the indications covered by marketed authorizations would confirm the appropriateness of selected dosages to the heterogeneous population actually treated.
Furthermore, some patients eligible for immunotherapies with immune checkpoint inhibitors do not respond to treatment. The heterogeneity of responses described by Chatterjee et al. [47] in their tumour growth model in advanced melanoma illustrates this well. The causes of these resistances to treatment are certainly multiple. Recent studies have highlighted, for example, the impact of digestive microbiota on the probability of the therapeutic success of immunotherapies [71].
Finally, beyond the choice of the right dose for a given population, the thorough elucidation of E–R relationships could open the way to personalized medicine. The study of ipilimumab [42] E–R relationships showed the correlation between exposure and OS, suggesting that ipilimumab therapeutic drug monitoring (TDM) would provide the means to achieve the best probability of survival. Indeed, clinical trials have shown that body weight-based dosing was not sufficient to control the PK variability of ipilimumab [31]. Two questions will need to be addressed before ipilimumab TDM can be made available to patients. First, it will be necessary to study the exposure–OS relationship before reaching steady state to determine whether early information on blood levels can guide dose selection. Since steady state is only reached after four courses of ipilimumab, it is too late to propose a dose adjustment. Second, it will be necessary to determine the optimal exposure that maximizes clinical efficacy. On the other hand, for pembrolizumab and nivolumab, the flat relationship observed between exposure and survival is in favour of higher-than-needed exposure [53, 56, 57]. Dose reduction is possible for these anti-PD1, but further studies will be needed to determine the minimum exposure required to reach the maximum efficiency plateau.
References
Eggermont AMM, Spatz A, Robert C. Cutaneous melanoma. Lancet. 2014;383:816–27.
Benlalam H, Labarrière N, Linard B, Derré L, Diez E, Pandolfino M-C, et al. Comprehensive analysis of the frequency of recognition of melanoma-associated antigen (MAA) by CD8 melanoma infiltrating lymphocytes (TIL): implications for immunotherapy. Eur J Immunol. 2001;31:2007–15.
Clemente CG, Mihm MC, Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303–10.
Nathanson L. Spontaneous regression of malignant melanoma: a review of the literature on incidence, clinical features, and possible mechanisms. Natl Cancer Inst Monogr. 1976;44:67–76.
Hua C, Boussemart L, Mateus C, Routier E, Boutros C, Cazenave H, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45–51.
Lerner AB. The Seiji memorial lecture. Pigment stories: from vitiligo to melanomas and points in between. Pigment Cell Res. 1992;Suppl 2:19–21.
Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–82.
Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–65.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;131:492–9.
Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–202.
Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–44.
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33.
Garbe C, Peris K, Hauschild A, Saiag P, Middleton M, Bastholt L, et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—update 2016. Eur J Cancer. 2016;63:201–17.
Schadendorf D, van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, et al. Melanoma. Lancet. 2018;392:971–84.
Napolitano S, Brancaccio G, Argenziano G, Martinelli E, Morgillo F, Ciardiello F, et al. It is finally time for adjuvant therapy in melanoma. Cancer Treat Rev. 2018;69:101–11.
Bristol-Myers Squibb Company. Ipilimumab (Yervoy). Highlights of prescribing information; 2018. https://packageinserts.bms.com/pi/pi_yervoy.pdf. Cited 18 Apr 2019.
Merck & Co., Inc. Pembrolizumab (Keytruda). Highlights of prescribing information; 2019. https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf. Cited 18 Apr 2019.
Bristol-Myers Squibb Company. Nivolumab (Opdivo). Highlights of prescribing information; 2019. https://packageinserts.bms.com/pi/pi_opdivo.pdf. Cited 18 Apr 2019.
Markham A, Duggan S. Cemiplimab: first global approval. Drugs. 2018;78:1841–6.
European Medicines Agency. Yervoy—European Public Assessment Report; 2011. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002213/WC500109302.pdf. Cited 27 Sep 2018.
Longoria TC, Tewari KS. Evaluation of the pharmacokinetics and metabolism of pembrolizumab in the treatment of melanoma. Expert Opin Drug Metab Toxicol. 2016;12:1247–53.
European Medicines Agency. Keytruda—European Public Assessment Report; 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003820/WC500190992.pdf. Cited 27 Sep 2018.
Wong ACY, Ma B. An update on the pharmacodynamics, pharmacokinetics, safety and clinical efficacy of nivolumab in the treatment of solid cancers. Expert Opin Drug Metab Toxicol. 2016;12:1255–61.
European Medicines Agency. Opdivo—European Public Assessment Report; 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003985/WC500189767.pdf. Cited 27 Sep 2018.
Shaabani S, Huizinga HPS, Butera R, Kouchi A, Guzik K, Magiera-Mularz K, et al. A patent review on PD-1/PD-L1 antagonists: small molecules, peptides, and macrocycles (2015–2018). Expert Opin Ther Pat. 2018;28:665–78.
Yang J, Hu L. Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: from antibodies to small molecules. Med Res Rev. 2019;39:265–301.
Oliva M, Rullan AJ, Piulats JM. Uveal melanoma as a target for immune-therapy. Ann Transl Med. 2016;4:172.
Berghoff AS, Venur VA, Preusser M, Ahluwalia MS. Immune checkpoint inhibitors in brain metastases: from biology to treatment. Am Soc Clin Oncol Educ Book. 2016;35:e116–22.
Cicchetti A, Coretti S, Mascia D, Mazzanti N, Refolo P, Rolli FR, et al. Assessing social and economic impact of subcutaneous mAbs in oncology. Glob Reg Health Technol Assess Ital North Eur Span. 2018;2018:1–9.
Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–64.
Feng Y, Masson E, Dai D, Parker SM, Berman D, Roy A. Model-based clinical pharmacology profiling of ipilimumab in patients with advanced melanoma. Br J Clin Pharmacol. 2014;78:106–17.
Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493–507.
Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. BioDrugs. 2010;24:23–39.
Ahamadi M, Freshwater T, Prohn M, Li C, de Alwis D, de Greef R, et al. Model-based characterization of the pharmacokinetics of pembrolizumab: a humanized anti-PD-1 monoclonal antibody in advanced solid tumors. CPT Pharmacomet Syst Pharmacol. 2017;6:49–57.
Elassaiss-Schaap J, Rossenu S, Lindauer A, Kang S, de Greef R, Sachs J, et al. Using model-based “learn and confirm” to reveal the pharmacokinetics–pharmacodynamics relationship of pembrolizumab in the KEYNOTE-001 trial. CPT Pharmacomet Syst Pharmacol. 2017;6:21–8.
Li H, Yu J, Liu C, Liu J, Subramaniam S, Zhao H, et al. Time dependent pharmacokinetics of pembrolizumab in patients with solid tumor and its correlation with best overall response. J Pharmacokinet Pharmacodyn. 2017;44:403–14.
Bajaj G, Wang X, Agrawal S, Gupta M, Roy A, Feng Y. Model-based population pharmacokinetic analysis of nivolumab in patients with solid tumors. CPT Pharmacomet Syst Pharmacol. 2017;6:58–66.
Liu C, Yu J, Li H, Liu J, Xu Y, Song P, et al. Association of time-varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin Pharmacol Ther. 2017;101:657–66.
Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9:204.
Maker AV, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, et al. Intrapatient dose escalation of anti-CTLA-4 antibody in patients with metastatic melanoma. J Immunother. 2006;29:455–63.
McDermott D, Haanen J, Chen T-T, Lorigan P, O’Day S, MDX010-20 Investigators. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20). Ann Oncol. 2013;24:2694–8.
Feng Y, Roy A, Masson E, Chen T-T, Humphrey R, Weber JS. Exposure–response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin Cancer Res. 2013;19:3977–86.
Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res. 2015;21:4286–93.
Sachs JR, Mayawala K, Gadamsetty S, Kang SP, de Alwis DP. Optimal dosing for targeted therapies in oncology: drug development cases leading by example. Clin Cancer Res. 2016;22:1318–24.
Lindauer A, Valiathan C, Mehta K, Sriram V, de Greef R, Elassaiss-Schaap J, et al. Translational pharmacokinetic/pharmacodynamic modeling of tumor growth inhibition supports dose-range selection of the anti-PD-1 antibody pembrolizumab. CPT Pharmacomet Syst Pharmacol. 2017;6:11–20.
Elassaiss-Schaap J. Allometric scaling in oncology disease progression from xenograft tumor growth to human non-small-cell lung cancer. In: 19th Annual Meeting of the Population Approach Group in Europe, 8–11 June 2010, Berlin.
Chatterjee M, Elassaiss-Schaap J, Lindauer A, Turner D, Sostelly A, Freshwater T, et al. Population pharmacokinetic/pharmacodynamic modeling of tumor size dynamics in pembrolizumab-treated advanced melanoma. CPT Pharmacomet Syst Pharmacol. 2017;6:29–39.
Claret L, Girard P, Hoff PM, Van Cutsem E, Zuideveld KP, Jorga K, et al. Model-based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J Clin Oncol. 2009;27:4103–8.
Goldmacher GV, Conklin J. The use of tumour volumetrics to assess response to therapy in anticancer clinical trials. Br J Clin Pharmacol. 2012;73:846–54.
Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908–18.
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–32.
Freshwater T, Kondic A, Ahamadi M, Li CH, de Greef R, de Alwis D, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43.
Turner D, Kondic AG, Anderson KM, Robinson A, Garon EB, Riess JW, et al. Pembrolizumab exposure–response assessments challenged by association of cancer cachexia and catabolic clearance. Clin Cancer Res. 2018;24(23):5841–9.
Agrawal S, Feng Y, Roy A, Kollia G, Lestini B. Nivolumab dose selection: challenges, opportunities, and lessons learned for cancer immunotherapy. J Immunother Cancer. 2016;4:72.
Wang Y, Sung C, Dartois C, Ramchandani R, Booth BP, Rock E, et al. Elucidation of relationship between tumor size and survival in non-small-cell lung cancer patients can aid early decision making in clinical drug development. Clin Pharmacol Ther. 2009;86:167–74.
Wang X, Feng Y, Bajaj G, Gupta M, Agrawal S, Yang A, et al. Quantitative characterization of the exposure–response relationship for cancer immunotherapy: a case study of nivolumab in patients with advanced melanoma. CPT Pharmacomet Syst Pharmacol. 2017;6:40–8.
Bajaj G, Gupta M, Feng Y, Statkevich P, Roy A. Exposure–response analysis of nivolumab in patients with previously treated or untreated advanced melanoma. J Clin Pharmacol. 2017;57(12):1527–33.
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–17.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34.
Long GV, Atkinson V, Cebon JS, Jameson MB, Fitzharris BM, McNeil CM, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202–10.
Lala M, Li M, Sinha V, de Alwis D, Chartash E, Jain L. A six-weekly (Q6W) dosing schedule for pembrolizumab based on an exposure–response (E–R) evaluation using modeling and simulation. J Clin Oncol. 2018;36(15 Suppl):3062.
Zhao X, Suryawanshi S, Hruska M, Feng Y, Wang X, Shen J, et al. Assessment of nivolumab benefit-risk profile of a 240-mg flat dose relative to a 3-mg/kg dosing regimen in patients with advanced tumors. Ann Oncol. 2017;28:2002–8.
Long GV, Tykodi SS, Schneider JG, Garbe C, Gravis G, Rashford M, et al. Assessment of nivolumab exposure and clinical safety of 480 mg every 4 weeks flat-dosing schedule in patients with cancer. Ann Oncol. 2018;29:2208–13.
Sandri M. Protein breakdown in cancer cachexia. Semin Cell Dev Biol. 2016;54:11–9.
Ternant D, Azzopardi N, Raoul W, Bejan-Angoulvant T, Paintaud G. Influence of antigen mass on the pharmacokinetics of therapeutic antibodies in humans. Clin Pharmacokinet. 2019;58:169–87.
Centanni M, Moes DJAR, Trocóniz IF, Ciccolini J, van Hasselt JGC. Clinical pharmacokinetics and pharmacodynamics of immune checkpoint inhibitors. Clin Pharmacokinet. 2019. https://doi.org/10.1007/s40262-019-00748-2.
Kaufman H, Schwartz LH, William WN, Sznol M, del Aguila M, Whittington C, et al. Evaluation of clinical endpoints as surrogates for overall survival in patients treated with immunotherapies. J Clin Oncol. 2017;35:e14557.
Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbe C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–20.
Hodi FS, Hwu W-J, Kefford R, Weber JS, Daud A, Hamid O, et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J Clin Oncol. 2016;34:1510–7.
Donia M, Kimper-Karl ML, Høyer KL, Bastholt L, Schmidt H, Svane IM. The majority of patients with metastatic melanoma are not represented in pivotal phase III immunotherapy trials. Eur J Cancer. 2017;74:89–95.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2017;359(6371):97–103.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Cyril Leven, Maël Padelli, Jean-Luc Carré and Eric Bellissant have no conflicts of interest to declare that are relevant to the content of this review. Laurent Misery has been a consultant for Sanofi, however there was no relationship with the studied drugs.
Funding
No sources of funding were used to assist in the preparation of this review.
Rights and permissions
About this article

Cite this article
Leven, C., Padelli, M., Carré, JL. et al. Immune Checkpoint Inhibitors in Melanoma: A Review of Pharmacokinetics and Exposure–Response Relationships. Clin Pharmacokinet 58, 1393–1405 (2019). https://doi.org/10.1007/s40262-019-00789-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00789-7