
Abstract
Background
Tildrakizumab is an anti-interleukin-23p19 monoclonal antibody recently approved for the treatment of chronic plaque psoriasis.
Methods
This analysis characterizes the population pharmacokinetics of subcutaneous tildrakizumab and identifies covariates influencing exposure in 2098 healthy volunteers and subjects with psoriasis. Tested covariates included body weight, formulation type, sex, age, race, serum albumin, creatinine clearance, Japanese origin, prior treatment with a biologic agent, subject status (subjects with psoriasis vs. healthy volunteers), and ethnicity.
Results
The pharmacokinetics was described by a one-compartment model with first-order absorption and elimination kinetics, and inter-individual variability on clearance, volume of distribution, and absorption rate constant. The pharmacokinetics was characterized by low clearance and limited volume of distribution. In subjects with psoriasis, the geometric mean clearance (coefficient of variation) was 0.32 L/day (38%), volume of distribution was 10.8 L (24%), and absorption and elimination half-life were 1.5 days (18%) and 23.4 days (23%), respectively, with an absorption lag time of 1.2 h. For the 100-mg dose, steady-state area under the plasma concentration vs. time curve for one dosing interval and maximum plasma concentration were 305 µg*day/mL (41%) and 8.1 µg/mL (34%), respectively. Steady state was achieved by 16 weeks with the clinical regimen (dosing on week 0 and week 4 and every 12 weeks thereafter) with 1.1-fold accumulation in maximum plasma concentration. Healthy subjects had 31% higher bioavailability than subjects with psoriasis. Subjects with increased body weight had a lower area under the plasma concentration-time curve at steady state vs. those with lower body weight. The modeled exposures were contained within clinical comparability bounds for all covariates including body weight.
Conclusions
The pharmacokinetics of tildrakizumab behaves like a typical monoclonal antibody without requiring dosage adjustment.
Trial Registration
NCT01729754, NCT01225731, NCT01722331.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Background
Psoriasis is a debilitating chronic inflammatory autoimmune skin disease influenced by both genetic and geographic predispositions [1, 2]. The current treatments for moderate-to-severe psoriasis include topical therapy and phototherapy, and small-molecule and biologic therapies. There is a significant unmet clinical need for novel effective psoriasis therapies owing to low patient satisfaction with existing therapies [3, 4].
Interleukin (IL)-23 has been identified as a key regulatory cytokine in the pathology of psoriasis responsible for stimulation of the differentiation, proliferation, and survival of T helper 17 cells [5, 6]. It is a heterodimer, and the first subunit (p40) is shared between IL-12 and IL-23, while the second subunit (p19) is specific to IL-23 alone [7, 8]. Specific blocking of IL-23, through the p19 subunit, has demonstrated important clinical improvement in the treatment of psoriasis [9, 10].
Tildrakizumab is a humanized immunoglobulin G1/κ anti-IL-23p19 monoclonal antibody that was recently approved for the treatment of moderate-to-severe chronic plaque psoriasis. Two pivotal, double-blind, randomized phase III studies evaluated the efficacy and safety of tildrakizumab relative to placebo and etanercept (reSURFACE 1 and reSURFACE 2, respectively) [10]. In these studies, tildrakizumab significantly improved skin clearance and patient Psoriasis Area Sensitivity Index 75 response relative to placebo and etanercept. Taken together, these results demonstrate the utility of monoclonal antibodies that selectively target IL-23p19 in the treatment of psoriasis.
The purpose of this analysis was to characterize the population pharmacokinetics of tildrakizumab and identify the intrinsic and extrinsic factors (covariates) influencing its exposure in healthy volunteers and subjects with psoriasis across three phase I trials (P05776, P06306, and P009), one phase IIb trial (P05495), and two phase III trials (P010 and P011). The model was then leveraged to support the assessment of clinical comparability bounds, clinical dose justification, and evaluation of the potential need for dose adjustments based on covariates.
2 Methods
2.1 Pharmacokinetic Studies
The population-pharmacokinetic (popPK) analysis was conducted on pharmacokinetic data obtained from a total of six studies (Table 1), including three phase I trials [P05776, P06306, and P009], one phase IIb trial [P05495 (NCT01225731)], and two phase III trials [P010 (NCT01722331) and P011 (NCT01729754)]. Two of the phase I trials were conducted in healthy subjects and were included in the popPK analysis because they contained rich profiles (including pharmacokinetic samples collected in the tildrakizumab absorption phase) and a wider dose range. The other studies were performed in patients with moderate-to-severe psoriasis. Only studies that included an active treatment arm with subcutaneous drug administration were used in this pooled data analysis. All trials were conducted in accordance with the guidelines on Good Clinical Practice and with the ethical standards for human experimentation established by the Declaration of Helsinki. All subjects provided written informed consent prior to participation in the individual trials. Further details on enrollment criteria and study inclusion/exclusion criteria can be found in the individual study publications [10,11,12,13,14].
2.2 Population-Pharmacokinetic Modeling Procedures
The popPK analysis was conducted using the non-linear mixed-effects modeling software NONMEM Version 7.3 (ICON Plc, Dublin, Ireland) running under PsN (Perl speaks NONMEM) 4.2.0 [15]. Model selection was based on NONMEM objective function value, goodness-of-fit plots, successful termination of the estimation and covariance routines, and the physiological plausibility and precision of the parameter estimates. The model was qualified for robustness and predictiveness with a non-parametric bootstrap [16] and a prediction-corrected visual predictive check [17], respectively. Serum tildrakizumab concentrations below the limit of quantification were treated as missing values and were not included in the analysis.
2.3 Analysis of Impact of Covariates on Pharmacokinetics
Continuous covariates of interest included baseline body weight (BW) (kg), age (years), serum albumin (g/L), and creatinine clearance (CrCL) calculated using the Cockroft–Gault formula (mL/min). Categorical covariates of interest included sex (male/female), race (White, Asian, other), ethnicity (non-Hispanic/Hispanic), Japanese origin (yes/no), previous therapy with biologic agents (yes/no), concomitant corticosteroid treatment (yes/no), formulation (lyophilized formulation from phase I and II studies vs. prefilled syringe from the phase III studies), and subject disease status (healthy/psoriatic). Missing covariates were indicated with a defined code (-99) in the data set. During the analysis, missing continuous covariate data were replaced by the median value for the respective covariate. Missing categorical values were replaced by the most frequent category of the respective covariate.
An automated forward selection and backward elimination process as implemented in the PsN stepwise covariate model (SCM) tool was used to identify significant covariates. In the forward step, covariates were added to the model one by one and retained if their inclusion constituted a significant improvement of the model fit. Each covariate effect retained in the forward step was subsequently eliminated from the model in a stepwise manner, unless removal resulted in a significant degradation in the model fit. The criterion for forward inclusion was p < 0.01 (change in objective function value of 6.63 for 1 degree of freedom). For backward elimination, a threshold of p < 0.001 (change in objective function value of 10.8 for 1 degree of freedom) was applied. Continuous covariates were included via a power function after centering on the median (parameter = typical parameter × [covariate/median covariate]power). For categorical covariates, a fractional change model was applied [parameter = typical parameter × [(1 + factor)].
2.4 Model Evaluation
Parameter precision for the popPK model was evaluated using the standard errors calculated from the NONMEM covariance step, as well as a non-parametric bootstrap analysis. A total of 1000 bootstrap replicates were generated by sampling randomly from the original data set with replacement. The resampling was stratified by trial to ensure a sufficient number of rich profiles in all the resampled data sets. Parameter estimates for each of the resampled data sets were obtained by fitting the final model. Non-parametric confidence intervals of the parameter estimates were derived. The final model was then evaluated by a simulation-based, prediction-corrected visual predictive check (as implemented in PsN).
2.5 Simulations for Univariate and Multivariate Analysis
The range of tildrakizumab exposures assumed to result in no clinically important change in safety or efficacy was derived by the observed exposure range across 100- and 200-mg dose regimens. The clinical comparability bounds relative to the clinical dose of 100 mg were determined to be 0.7–2.7. Clinical comparability was defined by the observation that no marked differences in efficacy (Psoriasis Area Sensitivity Index response) and safety (adverse events) were seen in exposure split out by quartile for 100 mg and 200 mg. Thus, bounds were defined by the median exposure of the extreme quartiles. The median exposure of the lowest exposure quartile (Q1) of patients receiving 100 mg of tildrakizumab every 12 weeks and the median exposure of the highest exposure quartile (Q4) of patients receiving 200 mg of tildrakizumab every 12 weeks were selected as boundaries. Average plasma concentration (Cavg) was chosen as the exposure metric; it was expressed in relation to the median exposure of patients receiving 100 mg of tildrakizumab every 12 weeks (marketed dose). Thus, the lower bound median was set as Cavg Q1 100 mg/median Cavg 100 mg (i.e., all patients receiving 100 mg) and the upper bound median was set as Cavg Q4 200 mg/median Cavg 100 mg. If exposures of different subgroups or exposure ranges related to observed ranges of continuous covariates fell within the clinical comparability bounds, no dose adaptation was deemed necessary.
The univariate effect of covariates on exposure [i.e., area under the plasma concentration-time curve at steady state (AUCss)] for 100 mg of tildrakizumab administered every 12 weeks was simulated by varying one covariate at a time while all other covariates were set to those of a typical subject. Exposures at the 5th and the 95th percentiles of the study population’s values for continuous covariates, and at the different levels of the categorical covariates, were compared with the exposure estimates for a typical subject and the range of exposures in the entire study population. The results were illustrated by a tornado plot.
To characterize differences in tildrakizumab pharmacokinetics between subpopulations (such as male/female, White/Asian), exposures were calculated using the pharmacokinetic fixed-effect parameters and the subject-specific covariates of all subjects receiving the 100-mg dose in a multivariate analysis. Geometric mean ratios were computed between the different subpopulations, and the results were illustrated by a forest plot. By using the observed covariate distributions, it was assured that correlations between covariates were taken into consideration.
3 Results
3.1 Data Description and Subject Characteristics
The full pharmacokinetic analysis data set consisted of 2106 subjects and 18,491 pharmacokinetic observations (Table 1). A total of 87 subjects were healthy volunteers and 2019 were patients with psoriasis. After exclusion of unevaluable data records (e.g., missing sampling times or concentrations), a total of 2098 subjects contributed 17,321 observation records. Of these, 3310 records were flagged as below the limit of quantification, and 44 outliers were identified by visual inspection for exclusion.
Subjects included in the final analysis dataset were predominantly Caucasian (80% of the overall population) and male (70.9% of the overall population), aged between 18 and 82 years (mean age 44.9 years), with BW ranging between 40.6 and 222.2 kg (mean BW 87.4 kg) and CrCL values between 33.3 and 395.7 mL/min (mean value 129.6 mL/min). Subject characteristics were generally similar across the studies except for a greater proportion of Asian subjects, and, consequently, a lower mean BW in Study P06306 (ethnic sensitivity study).
3.2 Pharmacokinetic Profile
The tildrakizumab concentration–time profiles stratified by dose for the phase I studies P05776, P06306, and P009 appeared to decline in a monophasic manner, indicating the pharmacokinetics could be described by a one-component structural model. There was no evidence of target-mediated drug disposition based on visual inspection of the concentration–time profiles. Dose-normalized concentrations were reasonably well super-imposed, suggesting tildrakizumab disposition was dose proportional within the range of tested doses (i.e., 5–400 mg).
The pharmacokinetic model was parameterized in terms of clearance (CL) and volume of distribution (V) with first-order absorption and elimination, and inter-individual variability on CL, V, and absorption rate constant. The residual error structure was a combined proportional and additive model.
A significant difference in pharmacokinetics between healthy subjects and subjects with psoriasis was observed in both the raw data and initial models. Including the covariate “subject status” on bioavailability (F) in the base model was necessary to obtain an acceptable fit. The difference between healthy subjects and subjects with psoriasis was partially explained by BW. Hence, BW was also included as a structural covariate on apparent CL and apparent V in the base model.
3.3 Covariate Analysis
In addition to the BW effect on apparent CL and apparent V and the subject status effect (healthy subject vs. psoriatic subject) on F, the following covariates were included in the SCM: age, sex, race, ethnicity, serum albumin, CrCL, previous use of biologic agents on apparent CL; age, sex, race, ethnicity, CrCL on apparent V; formulation on both absorption rate constant and F. Concomitant medication was not evaluable because of the low number of subjects taking systemic corticosteroids during a sufficiently long period (i.e., at least 8 weeks during the trial). In an alternative SCM, the covariate “race” (White/Asian/other) was exchanged for “Japanese” (yes/no). The SCM results are shown in Table 2 and were identical for both SCMs apart from race and Japanese not being tested in the respective other SCM.
3.4 Final Model
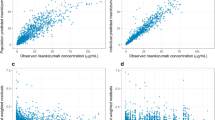
The final model adequately described the data (Fig. 1). Parameter estimates are reported in Table 3. The pharmacokinetic profile was characterized by low CL and limited V. In addition to the NONMEM-reported parameter estimates, geometric means and coefficient of variation were calculated for CL, V, absorption rate constant, and derived pharmacokinetic parameters. In subjects with psoriasis, the geometric mean CL (coefficient of variation) was 0.32 L/day (38%), V was 10.8 L (24%), and absorption and elimination half-life were 1.5 days (18%) and 23.4 days (23%), respectively, with an absorption lag time of 1.2 h. For both the 100- and 200-mg doses, time to maximum plasma concentration was 6.2 days (46%). Healthy subjects had 31% higher F than subjects with psoriasis. For the 100-mg dose, the steady-state area under the plasma concentration vs. time curve for one dosing interval and maximum plasma concentration were 305 µg*day/mL (41%) and 8.1 µg/mL (34%), respectively. The corresponding values for the 200-mg dose were 612 µg*day/mL (40%) and 16.3 µg/mL (33%), respectively. Steady state was achieved by 16 weeks with the clinical regimen (dosing on week 0 and week 4 and every 12 weeks thereafter) with 1.1-fold accumulation in maximum plasma concentration.

Goodness-of-fit plots for the final population-pharmacokinetic (PK) model for tildrakizumab. CWRES conditional weighted residuals, DV observation, IPRED individual predicted values, PRED predicted values, TAD time after dose. Note: Dots are individual data points; solid lines are smoothed loess lines. In the two plots of the first row, dashed lines are lines of identity, while in the two plots of the second row dashed lines show the boundaries of the CWRES ± 5 interval
3.5 Simulations Illustrating Magnitude of Covariate Effect
The isolated influence of each statistically significant covariate on the exposure of tildrakizumab after treatment with the 100-mg dose was evaluated in univariate simulations. The effects of each covariate on exposure were calculated one at a time, with all other covariates fixed to their typical values (as estimated for the reference subject). Continuous covariates were evaluated at the 5th and 95th percentiles of the entire trial population. Categorical covariate effects were evaluated for each level. Tornado plots of the effects of significant covariates on tildrakizumab AUCss are shown in Fig. 2. Prediction intervals for each of the covariates were within the observed clinical range.

Univariate impact of covariates on tildrakizumab area under the plasma concentration-time curve at steady state (AUCss) [100-mg dose administered every 12 weeks]. PI prediction interval
The range between the 5th and 95th percentiles of BW distribution in all subjects who received 100 mg of tildrakizumab (129–56 kg) correlated to a + 54% to − 34% change in tildrakizumab AUCss. Healthy subjects had a 31% higher exposure than a subject with psoriasis when all other covariates were held constant. Age resulted in + 9% and − 5% changes in exposure at the 5th and 95th percentiles of the age distribution. The range between the 5th and 95th percentiles of serum albumin distribution (40–50 g/L) correlated to a − 9% to + 9% change in exposure. Asian subjects had a 16% lower exposure compared with White subjects. Subjects of Hispanic ethnicity had a 15% lower exposure compared with non-Hispanic subjects. Women vs. men and non-Caucasians vs. Caucasians had 9% and 11% lower exposure, respectively. The lyophilized formulation used in early development had a 5% lower exposure than the prefilled syringe (no effect on absorption). Of note, the formulation difference was small relative to the healthy subject/subject with psoriasis difference. The 5th and 95th percentiles of AUCss for the total studied population that received 100 mg of tildrakizumab were 167 and 536 µg*day/mL, i.e., − 50% to + 59% difference from the typical value of 337 µg day/mL, respectively.
To evaluate the clinical relevance of intrinsic and extrinsic factors of clinical interest on the pharmacokinetics of tildrakizumab in the target population, individual model-predicted exposure estimates for subjects with psoriasis in the phase IIb and phase III trials who received 100 mg of tildrakizumab were stratified by covariates of clinical interest. Tildrakizumab exposure was summarized for demographic subgroups, and the geometric mean ratio of AUCss and the 90% confidence interval (CI) for comparisons between subgroups was calculated. The respective reference subgroups were: age < 65 years, BW ≤ 90 kg, normal albumin levels (35–50 g/L), male, White, non-Hispanic, and non-Japanese. The results were illustrated by a forest plot (Fig. 3).

Forest plot of geometric mean ratio (GMR) area under the plasma concentration-time curve at steady state (AUCss) [90% confidence interval (CI)] showing the multivariate impact of covariates on tildrakizumab exposure
Of all the covariates tested, BW had the most significant effect on AUCss (Fig. 3). The distribution of predicted AUCss values for 100- and 200-mg doses in subjects with psoriasis in different BW brackets were calculated (≤ 90 kg vs. > 90 kg) and displayed in box plots (Fig. 4). Steady-state area under the curve geometric mean ratios (90% CI) for > 90 kg/≤ 90 kg subjects receiving 100 mg and 200 mg subcutaneously were 0.71 (0.69–0.74) and 0.70 (0.67–0.72), respectively. The higher BW (> 90 kg) AUCss value was contained within the prespecified clinical comparability bounds for tildrakizumab, but near the lower bound of 0.7. The effects of all remaining covariates also fell within the clinical comparability bounds.

Tildrakizumab area under the plasma concentration-time curve at steady state (AUCss) plotted by dose and stratified by weight group (cut-off value: 90 kg)
4 Discussion
The popPK analysis was conducted based on six clinical studies in various phases of development including three phase I trials, one phase IIb trial, and two phase III trials. Two of the phase I trials were conducted in healthy subjects. These were included in the popPK analysis as they contained rich profiles (including pharmacokinetic samples collected in the tildrakizumab absorption phase), a wider dose range, and, in Trial P06306, a direct comparison between equal cohorts of White, Chinese, and Japanese subjects. Only active arms and arms with subcutaneous drug administration were used.
The serum tildrakizumab concentrations were well described by a one-compartment model with first-order absorption kinetics. As there was no evidence of target-mediated drug disposition based on visual inspection of individual concentration–time profiles, CL was described as linear first-order elimination. CL and V were in the expected range for therapeutic monoclonal antibodies.
Two covariates were found to substantially influence AUCss: subject status (healthy subjects vs. subjects with psoriasis) and BW. Owing to the imbalance in the number of subjects between the healthy category and the psoriasis category (i.e., only ~ 90 healthy subjects in the healthy volunteer trials P05776 and P06306 out of ~ 2100 subjects in total), it was not possible to obtain an unbiased model fit without using subject status as a covariate in the model. The covariate BW explained some of the discrepancy between healthy subjects and subjects with psoriasis, but not all. Inclusion of BW as a covariate reduced the difference in F between healthy subjects and subjects with psoriasis from 71 to 33%. Inclusion of all other covariates as described in the following paragraph only reduced the healthy vs. psoriatic difference in F by a further 2% to reach 31%.
Additional covariates were evaluated in a formal SCM procedure. Owing to the richness of the PK dataset, several covariates with small effects were identified as significant during the SCM: age, race (or alternatively, Japanese origin), ethnicity, sex, serum albumin, CrCL, and formulation. These covariates were associated with small effect sizes (race, ethnicity, age, sex, and formulation < 16% change in exposure). Covariates except BW and subject status had a very minor impact on diagnostic plots, and reductions in inter-individual variability were small (− 2% for inter-individual variability on CL and − 1% for inter-individual variability on V, comparing the base model with the final model). Residual errors remained unchanged.
The patient population with moderate-to-severe psoriasis is typically associated with a high BW. The median across phase IIb and III trials was found to be 86 kg with an extensive range from 41 to 222 kg. In the popPK analysis, BW was included as a covariate on CL and V as part of the structural model, as allometric principles typically apply for monoclonal antibodies. The popPK analysis demonstrated that drug CL and V increased with BW. Higher BW (> 90 kg) was associated with reduced exposure. Exposure for patients > 90 kg was found to be within the clinical comparability bounds, but near the lower bound. Hence, no dosage adjustment is recommended based on BW.
No other covariates were found to have a clinically relevant effect size on tildrakizumab exposure. Therefore, no dosage adjustment is warranted on the basis of sex, age, race, ethnicity, and Japanese origin.
Therapeutic antibodies are cleared by catabolism in a variety of tissues and are too large to pass through the glomerular basement membrane of the kidney [18, 19]. Therefore, no major changes in tildrakizumab exposure were anticipated in the setting of renal impairment. In the popPK model, CrCL was assessed for impact on tildrakizumab pharmacokinetics across phase I, IIb, and III trials, as it provides a good approximation of renal function. Most observed CrCL values were within the normal or mildly impaired renal function range. In the popPK model, CrCL was found to have a minor effect on tildrakizumab CL with the extremes of CrCL (range 33.26–395.73 mL/min) correlated to a − 16% to + 16% change in CL. The covariate effect is physiologically unlikely and more likely describing a correlative effect with age, sex, and BW, also included as covariates in the popPK model.
The mechanism of the relationship between albumin and CL of monoclonal antibodies has been described [20]. Albumin and antibodies are protected from lysosomal degradation by the same neonatal Fc receptor recycling system, although they interact with different binding sites. Factors that affect the capacity of neonatal Fc receptor recycling influence levels of albumin and immunoglobulin G antibodies in a similar manner; therefore, serum levels of albumin and immunoglobulin G antibodies increase and decrease concomitantly. Albumin was detected as a significant covariate in the popPK model but was found to not have a clinically significant impact on tildrakizumab exposure, as the magnitude of the effect fell within the clinical comparability bounds. Therefore, no dose adjustments are warranted on the basis of albumin. The normal range of serum albumin is defined as 35–50 g/L. It was not possible to assess the effect of low albumin levels on CL, as only two subjects in the 100-mg dose group and two subjects in the 200-mg dose group had serum albumin levels below the normal range. Hence, exposures of subjects in the normal range were compared to the few available subjects above the normal range (> 50 g/L).
Biologic agents, such as tildrakizumab, do not undergo typical metabolism or use transporter pathways that are relevant to small molecules. The lack of overlapping pathways is expected to limit the potential for pharmacokinetic-based interactions. Therefore, concomitant small-molecule medications are not anticipated to affect the pharmacokinetics of tildrakizumab. Concomitant use of systemic corticosteroids and prior treatment of psoriasis with a biologic agent were considered as potential covariates on the pharmacokinetics of tildrakizumab in the popPK analysis. However, concomitant corticosteroids could not be formally tested as a covariate in the popPK model because the predefined minimum of 50 subjects treated with systemic corticosteroids for at least 8 weeks was not met. Prior treatment of psoriasis with a biologic agent was tested but did not have a statistically significant influence on tildrakizumab exposure.
The effects of immunogenicity on tildrakizumab were summarized separately and not specifically assessed in the popPK model. It was observed that immunogenicity only had a modest impact on tildrakizumab pharmacokinetics. In subjects treated with either 100 or 200 mg tildrakizumab through weeks 52–64, approximately 7% developed antibodies to tildrakizumab and approximately 3% had neutralizing antibodies. Subjects who developed neutralizing antibodies showed a 36.5% increase in tildrakizumab median CL and reduced efficacy relative to subjects without antibodies [21].
5 Conclusions
These popPK findings indicate that the pharmacokinetics of tildrakizumab is characterized by low CL and limited V. Based on pharmacokinetic data only, there is no need for dosage adjustment for the tested intrinsic and extrinsic factors. BW had an effect on exposure, which, however, was contained within the clinical bounds for comparable efficacy and safety.
References
Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370(9583):263–71. https://doi.org/10.1016/S0140-6736(07)61128-3.
Parisi R, Symmons DP, Griffiths CE, Ashcroft DM. Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Investig Dermatol. 2013;133(2):377–85. https://doi.org/10.1038/jid.2012.339.
Duffin KC, Yeung H, Takeshita J, Krueger GG, Robertson AD, Troxel AB, et al. Patient satisfaction with treatments for moderate-to-severe plaque psoriasis in clinical practice. Br J Dermatol. 2014;170(3):672–80. https://doi.org/10.1111/bjd.12745.
Korman NJ, Zhao Y, Pike J, Roberts J, Sullivan E. Patient satisfaction with current psoriasis treatment: a real-world study in the USA. Dermatol Online J. 2016;22(2):1–10.
Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203(12):2577–87. https://doi.org/10.1084/jem.20060244.
Tonel G, Conrad C, Laggner U, Di Meglio P, Grys K, McClanahan TK, et al. Cutting edge: a critical functional role for IL-23 in psoriasis. J Immunol. 2010;185(10):5688–91. https://doi.org/10.4049/jimmunol.1001538.
Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140(3):645–53. https://doi.org/10.1016/j.jaci.2017.07.004.
Johnsson HJ, McInnes IB. Interleukin-12 and interleukin-23 inhibition in psoriatic arthritis. Clin Exp Rheumatol. 2015;33(5 Suppl. 93):S115–8.
Blauvelt A, Papp KA, Griffiths CE, Randazzo B, Wasfi Y, Shen YK, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76(3):405–17. https://doi.org/10.1016/j.jaad.2016.11.041.
Reich K, Papp KA, Blauvelt A, Tyring SK, Sinclair R, Thaci D, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390(10091):276–88. https://doi.org/10.1016/S0140-6736(17)31279-5.
Khalilieh S, Hodsman P, Xu C, Tzontcheva A, Glasgow S, Montgomery D. Pharmacokinetics of tildrakizumab (MK-3222), an anti-IL-23 monoclonal antibody, after intravenous or subcutaneous administration in healthy subjects. Basic Clin Pharmacol Toxicol. 2018;123(3):294–300. https://doi.org/10.1111/bcpt.13001.
Khalilieh S, Hussain A, Montgomery D, Levine V, Shaw PM, Bodrug I, et al. Effect of tildrakizumab (MK-3222), a high affinity, selective anti-IL23p19 monoclonal antibody, on cytochrome P450 metabolism in subjects with moderate to severe psoriasis. Br J Clin Pharmacol. 2018;84(10):2292–302. https://doi.org/10.1111/bcp.13670.
Papp K, Thaci D, Reich K, Riedl E, Langley RG, Krueger JG, et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol. 2015;173(4):930–9. https://doi.org/10.1111/bjd.13932.
Zandvliet A, Glasgow S, Horowitz A, Montgomery D, Marjason J, Mehta A, et al. Tildrakizumab, a novel anti-IL-23 monoclonal antibody, is unaffected by ethnic variability in Caucasian, Chinese, and Japanese subjects. Int J Clin Pharmacol Ther. 2015;53(2):139–46. https://doi.org/10.5414/CP202176.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit: a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57. https://doi.org/10.1016/j.cmpb.2005.04.005.
Efron B, Tibshirani RJ. Introduction to the bootstrap. Boca Raton: Chapman and Hall/CRC; 1993.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51. https://doi.org/10.1208/s12248-011-9255-z.
Meibohm B, Zhou H. Characterizing the impact of renal impairment on the clinical pharmacology of biologics. J Clin Pharmacol. 2012;52(1 Suppl.):54S–62S. https://doi.org/10.1177/0091270011413894.
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–58. https://doi.org/10.1038/clpt.2008.170.
Fasanmade AA, Adedokun OJ, Olson A, Strauss R, Davis HM. Serum albumin concentration: a predictive factor of infliximab pharmacokinetics and clinical response in patients with ulcerative colitis. Int J Clin Pharmacol Ther. 2010;48(5):297–308.
Kimball A, Blauvelt A, Reich K, Li Q, van Aarle F, Kerbusch T, et al. Immunogenicity with tildrakizumab, an anti-interleukin-23p19 monoclonal antibody, in a pooled analysis of three randomized controlled trials in patients with chronic plaque psoriasis. Br J Dermatol. 2017;177(5):E252–3.
Acknowledgements
The authors gratefully acknowledge the writing and editorial assistance of Amy O. Johnson-Levonas, Ph.D. and Anish Mehta (both of Merck & Co., Inc., Kenilworth, NJ, USA). The authors also acknowledge the assistance of Diana Montgomery (Merck & Co., Inc., Kenilworth, NJ, USA) in summarizing the immunogenicity data, and of Michele McColgan (Merck & Co., Inc., Kenilworth, NJ, USA) in formatting and preparing this article for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This analysis was supported by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and conducted by Certara USA Inc.
Conflict of interest
Petra Jauslin, Pooja Kulkarni, Hanbin Li, Suresh Vatakuti, and Thomas Kerbusch are employees of Certara USA, Inc. or its subsidiaries, who conducted analysis or provided consulting services for Merck & Co., Inc. Azher Hussain and Larissa Wenning are employees of Merck & Co., Inc., Kenilworth, NJ, USA, and hold stock and/or stock options in the company. Thomas Kerbusch is a former employee of Merck & Co., Inc., Kenilworth, NJ, USA, who was an employee of Certara USA, Inc. at the time of the analysis covered in this article.
Ethics approval
The studies used in this work were conducted in compliance with the Declaration of Helsinki, International Conference on Harmonization Guidelines for Good Clinical Practice, and applicable local regulations. The study protocols were approved by ethics committees of all participating centers.
Consent to participate
All patients provided written informed consent before study entry and the initiation of any study tests/procedures.
Rights and permissions
About this article

Cite this article
Jauslin, P., Kulkarni, P., Li, H. et al. Population-Pharmacokinetic Modeling of Tildrakizumab (MK-3222), an Anti-Interleukin-23-p19 Monoclonal Antibody, in Healthy Volunteers and Subjects with Psoriasis. Clin Pharmacokinet 58, 1059–1068 (2019). https://doi.org/10.1007/s40262-019-00743-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00743-7