
Abstract
Background and Objective
Leuprolide is a gonadotropin-releasing hormone (GnRH) agonist, which inhibits gonadotropin secretion by down-regulating pituitary GnRH receptor when administered continuously at therapeutic doses. The objectives of this analysis were to develop a population model that can describe the pharmacokinetics of the 6-month depot formulation of leuprolide acetate in patients with prostate cancer and to characterize the relationship of leuprolide plasma concentrations and serum testosterone concentrations.
Methods
The pharmacokinetic and pharmacodynamic analyses were performed using a non-linear mixed-effect modeling approach. Observations were pooled from studies on healthy male volunteers and prostate cancer patients, who were administered a single 1 mg intravenous dose of immediate-release leuprolide acetate and two intramuscular doses of 45 mg of the depot formulation, respectively. The covariates that were screened for the pharmacokinetic model included body weight, creatinine clearance, liver function markers (total bilirubin, blood urea nitrogen, AST, alanine aminotransferase), age, and body mass index.
Results
A two-compartment model with parallel first- and zero-order absorption processes and a delayed first-order process well-characterized the multi-phasic absorption profile of leuprolide acetate depot formulation. Typical population values of the absorption rate constant of the immediate and delayed processes were estimated to be 0.357 and 0.017 day−1, respectively, with a mean transit time of 9.5 days. No covariates were significant in this analysis. A semi-mechanistic model, which accounts for down-regulation of the GnRH receptor via an inhibitory maximum effect (E max) model and the stimulatory effect of activated receptors on testosterone levels, adequately described serum testosterone profiles following dosing. The equilibrium dissociation constant of leuprolide and the typical leuprolide plasma concentration required to achieve a castration testosterone level of ≤0.5 ng/mL were 0.3 and 0.03 ng/mL, respectively.
Conclusion
Population pharmacokinetics and pharmacodynamics of the leuprolide depot formulation were characterized using an integrated semi-mechanistic model. The developed model adequately describes the leuprolide–testosterone relationship and can potentially be used to facilitate design of clinical studies for new formulations, to aid in the selection of candidate formulations, and for the optimization of doses and dosing schemes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The pharmacokinetics of leuprolide acetate depot formulation were well-characterized using a two-compartment model with parallel first- and zero-order absorption processes and a delayed first-order absorption process. |
The effect of leuprolide on serum testosterone was described using a semi-mechanistic model, which accounts for down-regulation of the gonadotropin-releasing hormone receptor via an inhibitory maximum effect (E max) model and the stimulatory effect of activated receptors on testosterone levels. |
No subject covariates were found to affect the parameters of the leuprolide pharmacokinetic/pharmacodynamic model. |
1 Introduction
Prostate cancer is the second most common cancer and the second leading cause of cancer deaths in men in the USA. Approximately 233,000 new diagnoses and 29,480 prostate cancer deaths are estimated in 2014 [1]. For patients with metastatic or locally advanced prostate cancer, the standard treatment is androgen deprivation therapy (ADT) established surgically through bilateral orchiectomies or pharmacologically using gonadotropin-releasing hormone (GnRH) analogs. The latter is generally preferred due to its reversibility, ease of administration, and acceptability to patients [2–4]. The basis of ADT is to suppress serum testosterone levels to below the castration level, thereby inhibiting the growth of testosterone-dependent prostatic cancer cells.
Leuprolide (USAN) or leuprorelin (INN), a synthetic GnRH agonist with greater potency and a longer half-life than the endogenous GnRH, is used to treat a wide range of sex hormone-related disorders including advanced prostatic cancer, endometriosis, and central precocious puberty. It was first approved by the US FDA in 1985 for the palliative treatment of advanced prostate cancer. Sustained-release formulations (Lupron Depot®, AbbVie Inc., North Chicago, IL, USA) were subsequently developed to reduce the frequency of injections needed to maintain effective testosterone suppression [5–7]. With long-term exposure, leuprolide acts as a potent inhibitor of gonadotropin secretion by down-regulation of GnRH receptors in the pituitary, which results in suppressed circulating levels of luteinizing hormone (LH), follicle-stimulating hormone (FSH), and gonadal steroids after a transient hormonal flare [6]. Typically, within 4 weeks of leuprolide acetate therapy, serum testosterone decreases to near surgically castrated testosterone levels.
While a number of exposure-response and semi-mechanistic pharmacokinetic/pharmacodynamic models have been reported for GnRH antagonists, only a few have modeled the pharmacokinetic/pharmacodynamic relationship of GnRH agonists and serum testosterone [9–13]. No population pharmacokinetic or pharmacokinetic/pharmacodynamic model of leuprolide has been published to date. In this current work, the objectives are to explore the potential effects of patient demographics and covariates on leuprolide pharmacokinetics and to develop a population pharmacokinetic/pharmacodynamic model that incorporates receptor down-regulation in order to link the leuprolide exposure with the changes in testosterone levels after administration of leuprolide 6-month depot formulation.
2 Methods
2.1 Study Design and Subjects
A total of 151 patients with prostate cancer were enrolled in an open-label, multicenter phase III clinical trial. The study consisted of a screening period (up to 28 days), a 48-week treatment period, and a 30-day follow-up period. Subjects received two intramuscular (IM) injections of Lupron Depot® 45 mg 6-month formulation administered once every 24 weeks. The study drug was supplied in a prefilled, dual-chamber syringe that contained sterile lyophilized powder and diluent, which were mixed just before injection.
Male subjects, 18 years or older, with a pre-study serum testosterone level of >1.5 ng/mL and histologically confirmed prostate cancer (National Cancer Institute stages 2–4) or rising prostate-specific antigen following either radical prostatectomy (≤0.2 ng/mL increase from previous test on two consecutive assessments) or prostate irradiation (≤2.0 ng/mL increase above the nadir) were included in the study. Other study inclusion criteria comprised an Eastern Cooperative Oncology Group performance score of ≤2, a life expectancy of at least 18 months, and that at least 32 weeks had elapsed since prior hormone therapy [14].
For the characterization of the disposition of leuprolide, pharmacokinetic data following a single 1 mg intravenous (IV) bolus of immediate-release (IR) leuprolide acetate, available from a phase I study in six healthy volunteers, was also included [15]. Institutional review board approval was obtained at each site and written informed consent was provided by each subject before screening or any study-related procedures.
2.2 Sample Collection and Analytical Methods
In the phase III study, blood samples for pharmacokinetic and pharmacodynamic assessments were collected from a subset of 28 patients on study days 1, 2, 4, and 8, and at weeks 2, 4, 8, 14, 20, and 24 after the first IM administration. Similarly, samples were collected 1, 2, 4, and 8 days and 2, 4, 8, 14, 20, and 24 weeks from the second dose, i.e., study days 169, 170, 171, and 176 and at weeks 26, 30, 34, 40, 46, and 48. On study days 1 and 169, blood samples for pharmacokinetic assessment were collected pre-dose (0 h) and at 2, 4, and 8 h post-dose.
In the phase I study following the 1 mg IV dose, pharmacokinetic samples were collected from all subjects at pre-dose, 5, 10, 15, 20, 30, and 45 min and 1, 1.5, 2, 3, 4, 5, 6, 8, 12, and 24 h post-dose.
Leuprolide plasma concentrations were determined by a validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) method at PPD Development (Richmond, VA, USA), which has been previously reported [16]. The assay was selective for leuprolide with a lower limit of quantitation (LLOQ) of 0.025 ng/mL in 0.5 mL of plasma. Measurement of leuprolide plasma concentrations for the IV study was performed using a previously reported radioimmunoassay procedure with an LLOQ of 0.1 ng/mL [15].
Serum testosterone levels were measured in the phase III study using a validated liquid chromatography–mass spectrometry (LC–MS) method after non-polar solvent extraction at Esoterix (Calabasas Hills, CA, USA) and Abbott Bioanalysis (Abbott Park, IL, USA), as previously reported [17]. The LLOQ of this assay was 0.03 ng/mL.
2.3 Data Analysis
Non-linear mixed-effects modeling was performed using NONMEM® version 7.2 software (ICON Development Solutions, Ellicott City, MD, USA). The first-order conditional estimation method with interaction (FOCEI) was used. Diagnostic graphics and exploratory analyses were performed using R (version 3.1.0) and Xpose (version 4.3.2). The Perl-based software Perlspeaks-NONMEM (PsN) (version 3.6.2) was used to perform visual predictive checks.
The pharmacokinetic/pharmacodynamic data analysis was performed sequentially. Development of the population pharmacokinetic model started with construction of the base model, including the structural pharmacokinetic model and models for the inter-individual variability (IIV) and residual variability (includes within-individual variability, experimental errors, and/or model misspecifications, etc.). IIV was assumed to be log-normally distributed and modeled using an exponential error structure. Modeling of residual error using different error models (i.e., additive, proportional, and combined additive and proportional error) was explored. Once the base model was developed, the effect of covariates on the pharmacokinetic parameters was explored graphically and tested statistically using the forward inclusion and backward elimination approach. Individual posthoc pharmacokinetic parameters from the final pharmacokinetic model served as input for the pharmacodynamic model.
Leuprolide plasma concentration measurements below LLOQ (24 out of a total of 671 observations) were not included in the analysis.
2.4 Pharmacokinetic Model
2.4.1 Base Model
Disposition: As observed from the IV data, the leuprolide plasma concentration declined in a multi-exponential manner; hence, both two- and three-compartment models were fitted to the IV data. With the disposition model established, simultaneous fitting of IV and IM data were initially attempted. Due to a tendency for flip-flop kinetics of the individual profiles from the depot formulation study, population disposition parameters [clearance (CL), volume of distribution of the central compartment (V c), intercompartmental clearance (Q), and the volume of the peripheral compartment (V p)] were subsequently fixed to the values from the final IV pharmacokinetic model.
Absorption: Similar to other microsphere-based sustained-release formulations, leuprolide release from the 45 mg depot formulation is described as a multi-phase process with an initial ‘burst’ and a delayed slow release [6, 16, 18–20]. To describe this multi-phase absorption process, a number of different absorption models including first-order, zero-order and parallel first- and zero-order absorption together with a delayed first-order absorption process were explored. Delay was implemented using both a lag time and transit compartment model as previously reported by Savic et al. [21, 22]. The NONMEM® control stream file is provided in the Electronic Supplementary Material.
2.4.2 Covariate Model
Covariates screened for their possible effect on pharmacokinetic parameters included creatinine CL(CLCR), liver function markers (total bilirubin, blood urea nitrogen, AST, ALT), age, body weight, body surface area (BSA), and body mass index (BMI). CLCR and BSA were calculated using Cockcroft-Gault and Mosteller formulas, respectively [23, 24].
The impact of continuous covariates on the pharmacokinetic parameters was explored using power models, with the covariate scaled by the median value. Covariate modeling was performed using the forward inclusion [p < 0.01; objective function value (OFV) drop of at least 6.64 for 1 df], and backward-elimination approach (p < 0.001; OFV increase of 10.83 for 1 df) and was guided by evaluation of the empirical Bayes pharmacokinetic parameter estimates versus covariate plots as well as changes in the estimates of pharmacokinetic parameter variability and residual variability. Nested models were compared using the likelihood ratio test, while non-nested models were compared using the Akaike information criterion (AIC).
2.5 Pharmacodynamic Model
The model describing the relationship between pharmacokinetics and serum testosterone consisted of three sub-models, and was modified from the model described by Romero et al., for another GnRH agonist [8].
-
1.
Competitive interaction between endogenous GnRH and leuprolide for GnRH receptors was incorporated through Eq. 1:
$$ {\text{FRAC}} = \frac{{{\text{AGN}} + {\text{BGN}}}}{{{\text{AGN}} + {\text{BGN}} + 1}} $$(1)where FRAC is the fraction of activated receptor, AGN is the endogenous GnRH concentration normalized by its receptor equilibrium dissociation constant, and BGN is the leuprolide concentration normalized by its receptor equilibrium dissociation constant. At baseline, the fraction of activated receptor, FRAC0, is a function of \( \frac{\text{AGN}}{{1 + {\text{AGN}}}} \).
-
2.
The effect of leuprolide on receptor down-regulation was described by an indirect response model parameterized with a zero-order rate constant for the production (k SR) and a first-order rate constant for the degradation (k DR) of the total amount of receptor (RT). Inhibitory effect of leuprolide on the rate of production was modeled using an inhibitory maximum effect (E max) function and described by Eq. 2:
$$ \frac{\text{dRT}}{{{\text{d}}t}} = k_{\text{SR}} \times \frac{{\mathop {\text{DR}}\nolimits_{50}^{\gamma } }}{{\mathop {\text{DR}}\nolimits_{50}^{\gamma } + [{\text{FRAC}} - {\text{FRAC}}_{0} ]^{\gamma } }} - k_{\text{DR}} \times RT $$(2)where DR50 is the change in fraction of activated receptor from baseline needed to achieve half of the maximum synthesis rate and γ is the Hill’s constant, a shape factor. At baseline, the arbitrary total receptor amount, RT, is 1.
-
3.
The stimulatory effect of activated receptor on testosterone rate was modeled using Eq. 3:
$$ \frac{\text{dTestosterone}}{{{\text{d}}t}} = k_{\text{ST}} \times {\text{RT}} \times {\text{FRAC}} + k_{\text{IN}} - {\text{Testosterone}} \times k_{\text{DT}} $$(3)where k ST is the zero-order rate constant for the production of testosterone and k DT is the first-order rate constant for the degradation of testosterone. k IN is a zero-order input from non-LH/FSH-dependent testosterone production [25]. The total amount of activated receptor is a function of RT and the fraction of activated receptor. The significance of including the shape factor, γ, on the E max function and k IN were evaluated. We also explored the term RT/RT0 × (2 − RT/RT0), where RT0 is baseline total receptor, included by Romero et al. [8], to regulate the drop and recovery of RT, by taking into account the instantaneous amount of RT.
2.6 Model Assessment
Models were evaluated based on the goodness of fit to the data using the following criteria: (1) successful convergence of the estimation and covariance routines; (2) significant drop in the OFV as described earlier; (3) agreement in scatterplots of the population- and individual-predicted versus measured observations, and the lack of systematic patterns in scatterplots of conditional weighted residuals versus predicted observations and versus time; and (4) precision of the parameter estimates as assessed using the asymptotic standard errors obtained by the covariance routine in NONMEM®.
2.7 Model Qualification
Models during development were qualified by prediction-corrected visual predictive check (PC-VPC) where the final parameter estimates were used to simulate 1,000 replicates of the observed dataset [26]. Both observations and the simulated data were normalized on the typical model prediction for the median independent variable in each bin in order to account for variation in sampling times and predictive covariates introduced by binning of the observations. The median and 5th and 95th percentile concentrations of the simulated datasets were then plotted against the original observations.
3 Results
A total of 647 plasma leuprolide concentrations (101 from the IV study and 546 from the IM study) and 587 serum testosterone concentrations were analyzed. The demographic and clinical characteristics of the population included in the pharmacokinetic/pharmacodynamic analysis are summarized in Table 1.
3.1 Pharmacokinetic Model
A two-compartment disposition model with parallel first- and zero-order absorption processes characterizing the initial phase of drug release and a delayed first-order process characterizing the slower phase best described the leuprolide plasma concentration data from the depot formulation. The model was parameterized in terms of first-order absorption rate constants for the immediate and delayed absorption processes (k a2 and k a1, respectively), duration of infusion of the zero-order process (D3), mean transit time (MTT), number of transit compartments (N), absolute bioavailability (Fabs), CL, V c, Q, and V p.
Description of the disposition of the IV data using a three-compartment model was not found to provide a significantly better fit than a two-compartment model (p > 0.01), and this is consistent with previous reports [15]. Models of immediate zero- or first-order absorption followed by delayed first-order absorption models tended to produce poor prediction of concentrations after the first peak, particularly around the trough between the first and second peak. By allowing a combination of immediate zero- and first-order absorption and delayed first-order absorption, there was a significant improvement in the fit, with OFV improvement of more than 200. The duration of infusion for the zero-order process was estimated, using the data from the initial phase of drug release, to be 0.0095 days. The parameter was then fixed to this value in the simultaneous fitting of all data, which helped to improve model stability.
When covariates were added univariately in the forward inclusion process, only the effect of body weight on clearance (OFV drop of 12.37). However, there was no reduction in IIV, CL, or residual variability and the IIV of V c was inflated by twofold. As such, no covariates were retained in the final pharmacokinetic model, which included IIV on k a1, MTT, CL, and V c in addition to covariance between CL and V c. Attempts to include IIV on k a2 and inter-occasion variability of k a1 in the model were made but could not be well-estimated.
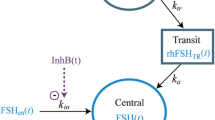
A schematic of the final structural pharmacokinetic model is illustrated in Fig. 1 and the parameter estimates, with the corresponding percentage relative standard errors (% RSE) are shown in Table 2. Goodness-of-fit to the observed concentrations is demonstrated in Fig. 2.

Schematic of pharmacokinetic/pharmacodynamic model structure. CL clearance, D3 duration of infusion for the zero-order absorption, F1 fraction of drug absorbed via delayed first-order process, F2 fraction of dose absorbed via immediate first-order process, F3 fraction of dose absorbed via zero-order process, GnRH gonadotropin-releasing hormone, k a1 absorption rate constant for delayed process, k a2 absorption rate constant for immediate process, k DR rate constant for degradation of total GnRH receptor, k DT rate constant for degradation of testosterone, k IN zero-order input from non-luteinizing hormone-/follicle-stimulating hormone-dependent testosterone production, k SR zero-order rate constant for the production of total GnRH receptor, k ST zero-order rate constant for the production of testosterone, k TR transit rate constant, Q inter-compartmental clearance

Goodness-of-fit plots for pharmacokinetics (left) and pharmacodynamics (right), with the top two rows presented on log scale. Circles represent the observations; the solid black and red lines show the line of identity and smoothed (loess) line through the data, respectively. CWRES conditional weighted residuals
Figure 3 shows the visual predictive check, where it can be observed that the 5th, 50th, and 95th percentiles of the prediction-corrected observations are in close agreement with the 95 % confidence intervals of their respective percentiles of the prediction-corrected simulated data, indicating ability of the selected model to describe the central tendency and variability of the observed data.

Prediction-corrected visual predictive check for the pharmacokinetic model. Leuprolide concentrations are on the log-scale. Circles represent prediction-corrected observations; red lines represent 5th (dashed), 50th (solid), and 95th (dashed) percentiles of the prediction-corrected observations; and shaded areas represent 95 % confidence intervals of the 5th, 50th, and 95th percentiles of prediction-corrected simulated data
3.2 Pharmacodynamic Model
Figure 1 shows the schematic representation of the final population pharmacokinetic/pharmacodynamic model of the effect of leuprolide on serum testosterone levels for patients with prostate cancer. The parameter estimates, with the corresponding % RSE, are summarized in Table 3. The goodness-of-fit plots show that the final model predicts the observed testosterone concentrations adequately, with no indication of systematic bias (Fig. 2). In this final pharmacodynamic model, only IIV on baseline testosterone could be supported by the data. Inclusions of IIV on other parameters were attempted but were either not successful or could not be precisely estimated. Addition of the term RT/RT0 × (2 − RT/RT0), which was included by Romero et al. [8] to regulate decrease and recovery of RT, was not found to significantly improve the fit and could not be estimated together with the Hill’s constant. Inclusion of the Hill’s constant allowed for flexibility in the degree of sigmoidicity in the relationship between the down-regulation effect and change in the fraction of activated receptor from baseline. This latter model produced similar population estimates as the model with the RT/RT0 × (2 − RT/RT0) term with better precision and slightly improved AIC.
By solving the differential equations using estimates of the typical values from the model, and assuming a target castration testosterone level of 0.5 ng/mL [27], the typical leuprolide plasma concentration needed to achieve the target testosterone level was estimated to be 0.03 ng/mL. Figure 4 shows the visual predictive check for the pharmacodynamic model, where it can be observed that the 5th, 50th, and 95th percentiles of the prediction-corrected testosterone levels are in close agreement with the 95 % confidence intervals of their respective percentiles of the prediction-corrected simulated data, indicating ability of the selected model to describe the central tendency and variability of the observed testosterone concentrations.

Prediction-corrected visual predictive check for the pharmacodynamic model. Testosterone concentrations are on the log-scale. Circles represent the prediction-corrected observations; red lines represent 5th (dashed), 50th (solid), and 95th (dashed) percentiles of the prediction-corrected observations; and shaded areas represent 95 % confidence intervals of the 5th, 50th, and 95th percentiles of prediction-corrected simulated data
4 Discussion
To the best of our knowledge, this is the first report of the development of a population pharmacokinetic or a population pharmacokinetic/pharmacodynamic model for leuprolide. The models were developed using pharmacokinetic and pharmacodynamic data from a multicenter, phase III clinical trial where patients with prostate cancer were treated with leuprolide 6-month depot formulation for 48 weeks and pharmacokinetic data from healthy volunteers who received the IR leuprolide 1 mg formulation.
Leuprolide plasma concentration–time profiles were well-described using a two-compartment disposition model with parallel first- and zero-order absorption and a delayed first-order absorption process. Typical of microsphere depot formulations, the pharmacokinetic profile of leuprolide 6-month depot formulation exhibited an initial burst phase followed by a later prolonged-release phase. This initial rapid rise in plasma concentration is often attributed to a combination of release of the drug from the swelled polymer matrix near the surface and through the hydrated aqueous channels produced by the connected drug cores [20, 28, 29]. The delayed phase of release results from the slow erosion of the core of the biodegradable polymer. For the modeling of this delayed-phase, transit compartment absorption model, which describes drug absorption as a multiple step process represented by a chain of pre-systemic compartments, was preferred over a lag-time model. The advantage of the absorption transit model is that it allows for a more gradual change, and hence describes the delay in a more physiologically plausible manner. Furthermore, unlike the lag-time model, it is more stable and IIV of the delay could be estimated.
In this analysis, the population estimate of leuprolide CL is 198 L/day, which is consistent with the previously reported mean values of 166–271 L/day in patients with prostate cancer [30], and mean values of 218 and 199 L/h after IV and subcutaneous (SC) administration of IR leuprolide, respectively, in healthy subjects [15]. While an assumption that leuprolide disposition in healthy subjects and patients with prostate cancer is similar was made for the analysis, this is supported by the lack of appreciable disposition differences in these two populations as observed in previous reports. Besides comparable CLs, the mean volume of distribution at steadystate (37.1 L) after SC administration of IR leuprolide 1 mg in healthy subjects (n = 6) was also similar to that in prostate cancer patients (n = 11 to n = 6), where the mean ranged from 27.3 to 36 L [18, 30].
For populations with severe renal or hepatic impairment, leuprolide pharmacokinetics have not been formally characterized. In our analysis, where the majority of the patients have mild or moderately impaired renal function, a trend towards reduction in leuprolide CL with decrease in CLCR was observed but the association was not statistically significant (p > 0.05). No association between hepatic function biomarkers (ALT, AST, and total bilirubin) and leuprolide CL was observed. Whilst the modest number of subjects with hepatic impairment or severe renal impairment may have limited the power to detect significance, it is important to note that for sustained-release formulations, drug concentration–time profiles are governed mainly by the slow release; hence, alterations in disposition with renal or hepatic function would not be expected to have a major influence. Furthermore, with the wide safety margin of leuprolide and the established safety profile in large patient populations, where mild to moderate renal impairment pre-exists, there is no evidence of a need for dose adjustment in patients with renal or hepatic impairment. This is consistent with the lack of recommendation for dose adjustments for depot formulations of the other GnRH agonists, triptorelin and goserelin in patients with renal or hepatic impairment. This is despite the fact that triptorelin CL following 0.5 mg IV bolus administration was reported to decrease with decreasing renal and hepatic function [31]. As for goserelin, decreased CLs were found to be correlated to reductions in renal function but not hepatic impairment [32].
There has been no evidence in the literature of an effect of BMI on the pharmacokinetics of leuprolide or consensus on its relationship with the outcome of testosterone suppression. In one study conducted in 49 prostate cancer patients, it was observed that men with a BMI >30 kg/m2 had higher total and free testosterone levels during leuprolide 3-month depot treatment than men with a normal BMI [33]. Other studies, however, have not noted differences in serum testosterone concentration among normal weight, overweight, and obese patients receiving GnRH agonist therapy for prostate cancer [34–36]. Furthermore, in the larger pivotal clinical study of the 6-month depot formulation (n = 150) [14], it was observed that serum testosterone was suppressed to values ≤0.5 ng/mL from week 4 through week 48 at a similar rate for the subgroups based on BMI. We explored the relationship between BMI on the absorption parameters, and found no evidence of its effect on either the absorption rate and MTT of the delayed absorption process. This seems to be consistent with the observation that absorption following IM administration is generally less susceptible to differences in body composition, such as in the case of obesity or sex difference, than SC administration. It should be noted that the small proportion of obese patients (14 %) may be a limitation. Similarly, no effect of body weight on CL has been observed. In this analysis, there was a similar weight distribution between the healthy and patient populations (84 ± 18 vs. 84 ± 14 kg, respectively).
Based on the framework of the receptor down-regulation model proposed by Romero et al. [8], the current model was able to adequately describe serum testosterone profiles following leuprolide dosing. Indirect response models were used to characterize leuprolide effect on receptor dynamics, and the stimulatory effect of activated receptors on testosterone production in this analysis. Estimated values for system parameters such as k DT and k IN are 0.01 h−1 and 0.018 ng/mL/day, respectively. These are similar to the values published previously for k DT and k IN [8, 12, 37]. At baseline, production of testosterone via the process independent of GnRH receptor activation is estimated to be approximately 0.37 % of the total production rate of testosterone, which is in close agreement to the value of 0.43 % reported previously [8]. This contribution to the overall production of testosterone might be representative of the synthesis of testosterone from dehydroepiandrosterone in the general circulation [25]. Estimate of k d, the equilibrium binding constant of leuprolide, is within the range reported from in vitro binding assays [38]. A limitation of this model is the inability to estimate IIV of parameters other than baseline testosterone, and this could be in part due to the lack of data from different dose levels. Nonetheless, the work presented serves as a good framework and was able to describe the pharmacokinetic/pharmacodynamic relationship for leuprolide without the complexity of modeling the dynamics of the hypothalamus–pituitary gland axis, including LH and precursor pool of LH as well as involvement of multiple feedback interactions, which often results in long run times [12, 13].
In order to establish a more direct relationship of leuprolide concentrations to the outcome of testosterone concentrations in this complex system, we calculated the minimum leuprolide concentration needed to achieve and maintain the castration testosterone level of 0.5 ng/mL. This was based on the typical values from the established population pharmacodynamic model. While there have not been any previous reports for leuprolide minimum effective concentration, the concentration of 0.03 ng/mL estimated in our analysis is close to the estimated median value of 0.0356 ng/mL required to achieve the castration testosterone level [8] and the EC50 (concentration of drug producing 50 % of E max) estimate of 0.047 ng/mL reported for another agonist with comparable potency [12]. This finding is supported by two pharmacokinetic/pharmacodynamic studies of the 1- and 3-month leuprolide depot formulations, where castration testosterone levels (≤0.5 ng/mL) were maintained while leuprolide concentrations in the prolonged-release phase were observed to be above 0.1 ng/mL [26, 39]. Our findings could be further validated through pharmacokinetic evaluation of patients who fail to achieve a castration testosterone level.
5 Conclusion
Good characterization of the pharmacokinetics of the 6-month depot formulation of leuprolide was achieved using non-linear mixed-effects modeling. No clinically relevant relationship with the demographic covariates tested was found in the absorption and disposition properties. A pharmacokinetic/pharmacodynamic model was also developed and adequately described the effect of leuprolide on the dynamics of testosterone via receptor down-regulation. This established model can potentially be used for optimization of doses and dosing schemes, as well as to facilitate selection of new candidate formulations, hence allowing for a more rational and model-based drug development.
References
American Cancer Society. What are the key statistics about prostate cancer? 2014. http://www.cancer.org/cancer/prostatecancer/detailedguide/prostate-cancer-key-statistics. Accessed 8 Nov 2014.
National Cancer Institute. Prostate cancer treatment (PDQ®). 2014. http://www.cancer.gov/cancertopics/pdq/treatment/prostate/HealthProfessional/page7. Accessed 8 Nov 2014.
Saylor PJ, Smith MR. Adverse effects of androgen deprivation therapy: defining the problem and promoting health among men with prostate cancer. J Natl Compr Canc Netw. 2010;8:211–23.
Trachtenberg J. Hormonal management of stage D carcinoma of the prostate. In: Carson CC, editor. Problems in urology, vol. 7. Philadelphia: JB Lippincott Co; 1993. p. 215–25.
Perrin MH, Rivier JE, Vale WW. Radioligand assay for gonadotropin-releasing hormone: relative potencies of agonists and antagonists. Endocrinology. 1980;106:1289–93.
US Food and Drug Administration. Lupron Depot label information. 2014. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.DrugDetails. Accessed 8 Nov 2014.
Conn PM, Roger SC, Seay SG. Biphasic regulation of gonadotropin-releasing hormone receptor by receptor microaggregation and intracellular Ca2+ levels. Med Pharmacol. 1984;25:51–5.
Romero E, Vélez de Mendizabal N, Cendrós J-M, Peraire C, Bascompta E, Obach R, et al. Pharmacokinetic/pharmacodynamic model of the testosterone effects of triptorelin administered in sustained release formulations in patients with prostate cancer. J Pharmacol Exp Ther. 2012;342(3):788–98.
Fattinger KE, Verotta D, Porchet HC, Munafo A, Le Cotonnec JY, Sheiner LB. Modeling a bivariate control system: LH and testosterone response to the GnRH antagonist antide. Am J Physiol. 1996;271(4 Pt 1):E775–87.
Pechstein B, Nagaraja NV, Hermann R, Romeis P, Locher M, Derendorf H. Pharmacokinetic-pharmacodynamic modeling of testosterone and luteinizing hormone suppression by cetrorelix in healthy volunteers. J Clin Pharmacol. 2000;40(3):266–74.
Wong SL, Lau DT-W, Baughman SA, Fotheringham N, Menchaca D, Garnick MB. Pharmacokinetics and pharmacodynamics of a novel depot formulation of abarelix, a gonadotropin-releasing hormone (GnRH) antagonist, in healthy men ages 50 to 75. J Clin Pharmacol. 2004;44(5):495–502.
Tornoe CW, Agers H, Senderovitz T, Nielsen HA, Madsen H, Karlsson MO, et al. Population pharmacokinetic/pharmacodynamic (PK/PD) modeling of the hypothalamic-pituitary-gonadal axis following treatment with GnRH analogues. Br J Clin Pharmacol. 2007;63(6):648–64.
Jadhav PR, Agersø H, Tornøe CW, Gobburu JVS. Semi-mechanistic pharmacodynamic modeling for degarelix, a novel gonadotropin releasing hormone (GnRH) blocker. J Pharmacokinet Pharmacodyn. 2006;33(5):609–34.
Spitz A, Young JM, Larsen L, Mattia-Goldberg C, Donnelly J, Chwalisz K. Efficacy and safety of leuprolide acetate 6-month depot for suppression of testosterone in patients with prostate cancer. Prostate Cancer Prostatic Dis. 2012;15(1):93–9.
Sennello LT, Finley RA, Chu SY, Jagst C, Max D, Rollins DE, et al. Single-dose pharmacokinetics of leuprolide in humans following intravenous and subcutaneous administration. J Pharm Sci. 1986;75(2):158–60.
Mostafa NM, Chwalisz K, Larsen L, Mattia-Goldberg C, Spitz A, Pradhan RS. Evaluation of the pharmacokinetics and pharmacodynamics of two leuprolide acetate 45 mg 6-month depot formulations in patients with prostate cancer. Clin Pharmacol Drug Dev. 2014;3(4):270–5.
Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab. 2007;92:405–13.
Periti P, Mazzei T, Mini E. Clinical pharmacokinetics of depot leuprorelin. Clin Pharmacokinet. 2002;41(7):485–504.
Okada H, Inoue Y, Heya T, Ueno H, Ogawa Y, Toguchi H. Pharmacokinetics of once-a-month injectable microspheres of leuprolide acetate. Pharm Res. 1991;8(6):787–91.
Okada H, Doken Y, Ogawa Y, Toguchi H. Preparation of three-month depot injectable microspheres of leuprorelin acetate using biodegradable polymers. Pharm Res. 1994;11(8):1143–7.
Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34(5):711–26.
Wilkins JJ, Savic RM, Karlsson MO, Langdon G, McIlleron H, Pillai G, et al. Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob Agents Chemother. 2008;52(6):2138–48.
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41.
Mosteller R. Simplified calculation of body-surface area. N Engl J Med. 1987;317(17):1098.
Labrie F. Hormonal therapy of prostate cancer. Prog Brain Res. 2010;182:321–41.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51.
Zlotta A, Debruyne FMJ. Expert opinion on optimal testosterone control in prostate cancer. Eur Urol Suppl. 2005;4:37–41.
Okada H. One- and three-month release injectable microspheres of the LH–RH superagonist leuprorelin acetate. Adv Drug Deliv Rev. 1997;28(1):43–70.
Gombotz WR, Pettit DK. Biodegradable polymers for protein and peptide drug delivery. Bioconjug Chem. 1995;6(4):332–51.
Mazzei T, Mini E, Rizzo M, Periti P. Human pharmacokinetic and pharmacodynamic profiles of leuprorelin acetate depot in prostatic cancer patients. J Int Med Res. 1990;18(Suppl 1):42–56.
Muller FO, Terblanche J, Schall R, van Zyl Smit R, Tucker T, Marais K, et al. Pharmacokinetics of triptorelin after intravenous bolus administration in healthy males and in males with renal or hepatic insufficiency. Br J Clin Pharmacol. 1997;44(4):335–41.
Cockshott ID. Clinical pharmacokinetics of goserelin. Clin Pharmacokinet. 2000;39(1):27–48.
Smith MR. Obesity and sex steroids during gonadotropin-releasing hormone agonist treatment for prostate cancer. Clin Cancer Res. 2007;13(1):241–5.
Fujii Y, Yonese J, Kawakami S, Yamamoto S, Okubo Y, Fukui I. Equivalent and sufficient effects of leuprolide acetate and goserelin acetate to suppress serum testosterone levels in patients with prostate cancer. BJU Int. 2008;101(9):1096–100.
Lee S-H, Lee H-M, Kim S-W, Lee E-S, Hong S-J, Kim C-S, et al. Is high-dose leuprorelin acetate effective and safe in Asian men with prostate cancer? An open-label, non-comparative, multi-center clinical trial. Yonsei Med J. 2014;55(2):310.
Van der Sluis TM, van Moorselaar RJA, Meuleman EJH, ter Haar RW, Bui HN, Heijboer AC, et al. Relationship between body mass index and serum testosterone concentration in patients receiving luteinizing hormone-releasing hormone agonist therapy for prostate cancer. Urology. 2013;81(5):1005–9.
Gries JM, Munafo A, Porchet HC, Verotta D. Down-regulation models and modeling of testosterone production induced by recombinant human choriogonadotropin. J Pharmacol Exp Ther. 1999;289(1):371–7.
Struthers RS, Xie Q, Sullivan SK, Reinhart GH, Kohout TA, Zhu YF, et al. Pharmacological characterization of a novel nonpeptide antagonist of the human gonadotropin-releasing hormone receptor, NBI-42902. Endocrinology. 2007;148(2):857–67.
Khan MS, O’Brien A. An evaluation of pharmacokinetics and pharmacodynamics of leuprorelin acetate 3M-depot in patients with advanced and metastatic carcinoma of the prostate. Urol Int. 1998;60(1):33–40.
Disclosure statement
The design, study conduct, and financial support for the clinical trials were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication. Both authors are employees of AbbVie and have no additional conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lim, C.N., Salem, A.H. A Semi-Mechanistic Integrated Pharmacokinetic/Pharmacodynamic Model of the Testosterone Effects of the Gonadotropin-Releasing Hormone Agonist Leuprolide in Prostate Cancer Patients. Clin Pharmacokinet 54, 963–973 (2015). https://doi.org/10.1007/s40262-015-0251-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0251-9