
Abstract
Background and Objectives
In clinical practice, antiretroviral regimens are often interrupted or modified for intolerance and toxicity. The objective of this study was to develop an in vitro to in vivo extrapolation (IVIVE) approach to describe the interaction when efavirenz is switched to either maraviroc or nevirapine and to test different switching scenarios to identify the best strategy.
Methods
In vitro data describing the chemical and absorption, tissue distribution, metabolism and excretion (ADME) characteristics of efavirenz, maraviroc and nevirapine were obtained from the literature, and used to simulate plasma exposures of these drugs using the Simcyp Population-Based Simulator. The predicted maraviroc and nevirapine exposures were compared with data from clinical studies evaluating their exposures following a switch from efavirenz.
Results
Model predictions for maraviroc and nevirapine exposure were in agreement with observed data. The simulations suggest that the waning efavirenz induction effect following discontinuation necessitated increasing maraviroc to 600 mg twice daily for 1 week after efavirenz cessation. Alternatively, adequate exposure of maraviroc was shown with a dose of 450 mg for 2 weeks. Efavirenz waning induction did not affect nevirapine exposure.
Conclusion
IVIVE modelling successfully predicted patient drug exposure. This modelling technique is able to inform the design of clinical studies, and allows assessment of pragmatic dosing strategies under complex therapeutic scenarios.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
An in vitro to in vivo extrapolation model to describe antiretroviral drug–drug interactions has been developed and validated. |
The model has the ability to inform the design of clinical studies. |
The model can also be used for the assessment of pragmatic dosing strategies under complex therapeutic scenarios. |
1 Introduction
Efavirenz-based antiretroviral combinations are first-line regimens for treatment of naive HIV-1 patients. Central nervous system (CNS) adverse events are common following initiation with efavirenz, often necessitating a switch to alternative agents in patients with ongoing CNS toxicity [1–3]. In these circumstances it is important to manage drug interactions leading to low concentrations of the alternative agent due to persisting enzyme induction by efavirenz, to avoid allowing a window for virological replication and escape.
Efavirenz is mainly metabolised by the cytochrome P450 (CYP) 2B6 isoform, and secondarily by CYP3A4, CYP2A6, CYP1A2 and uridine-5′-diphospho-glucuronosyltransferase (UGT) 2B7 [4, 5]. In vivo, efavirenz causes induction of CYP3A4 and CYP2B6, although the magnitude of an interaction is related to the degree of induction [6]. Moreover, concomitant drugs inducing or inhibiting CYP2B6 and CYP3A4 can affect efavirenz metabolism and exposure [5].
Maraviroc is a selective C-C chemokine receptor type 5 (CCR5) antagonist, inhibiting the interaction of HIV-1 gp120 and CCR5 that is necessary for CCR5-tropic HIV-1 to enter cells [7]. Maraviroc is extensively metabolised by CYP3A4; thus, its exposure is altered by concomitant drugs that modulate activity of CYP3A4 and maraviroc dose adjustment might be necessary [8]. The maraviroc dose should be increased from 300 mg twice daily to 600 mg twice daily when co-administered with efavirenz. Due to the long efavirenz half-life (40–55 h, or longer at steady state) [4, 9], CYP3A4 induction can be prolonged for many days, even after cessation; consequently, efavirenz may affect maraviroc concentrations during the period of switching. The length of time for which efavirenz can induce drug metabolism has not been fully characterised and can be affected by many factors (e.g. demographic, diseases status, genetics). Consequently, choosing the appropriate switching strategy is crucial. Starting maraviroc therapy immediately after efavirenz cessation may result in sub-therapeutic maraviroc concentrations with possible reduced efficacy, and keeping an increased maraviroc dose for an excessive length of time may result in concentration-dependent adverse events (i.e. hypotension) [10].
Nevirapine is mainly metabolised in the liver by CYP3A4 and CYP2B6 [11], inducing its own metabolism and resulting in a 1.5- to 2-fold increase in the oral clearance after the first weeks of dosing [12]. For this reason, lead-in dosing of nevirapine (200 mg for 14 days, increasing to 200 mg twice daily thereafter in adults) is recommended, but it is unclear whether such dosing results in a window of sub-therapeutic nevirapine concentrations following a switch from efavirenz to nevirapine [13–17].
In vitro to in vitro extrapolation (IVIVE) is a ‘bottom-up’ approach that integrates drug-specific factors, human physiology and anatomy through mathematical modelling, to simulate pharmacokinetics in virtual populations. Recent US Food and Drug Administration guidance on metabolic drug–drug interactions (DDIs) has put more emphasis on the use of in vitro systems for detecting and anticipating such effects [18]. The in vitro detection of potential DDIs has been extensively reviewed. DDIs represent a relevant medical problem with economic loss for the health system, and early prediction and assessment of potentially significant DDIs that may result in adverse effects is extremely important. The objective of this study was to develop an IVIVE modelling approach to test different switching scenarios away from efavirenz. Here we describe the switch to maraviroc or nevirapine since we were able to compare modelling data with clinically derived pharmacokinetic parameters. We have identified the best strategies for each switch.
2 Methods
2.1 Drug of Parameters
The oral absorption, tissue distribution, metabolism and excretion (ADME) of each drug were simulated using the Simcyp Population-Based Simulator (version 11; Simcyp, Sheffield, UK) in healthy subjects. The Simcyp program creates virtual populations with genetic, physiological and demographic variables that are generated using equations derived from population databases obtained from literature sources. The relevant parameters to the IVIVE scaling process are obtained for each individual and are then applied, together with in vitro metabolism data, to obtain whole liver intrinsic clearance values [19]. In vitro parameters were scaled to hepatic and intestinal unbound intrinsic clearance (CLuH,int and CLuG,int for liver and gut, respectively) via incorporation of covariate-linked scaling factors generated for each virtual subject as described previously [20]. Hepatic clearance (CLH), fraction escaping the first-pass metabolism (F H) and fraction escaping the gut metabolism (F G) were then calculated, incorporating additional individualised system parameters of liver blood flow, free fraction in blood (fuB) and enterocytes (fuGUT), and blood flow in the gut (Q GUT) as reported previously [20]. Simcyp simulates a virtual population that, ideally, captures the range of inter-subject variability of real patient populations. Using in vitro data on drug metabolism and incorporating inter-individual variability that is relevant to drug metabolism in both the liver and gut, the Simcyp algorithms have been used to predict the clearances of the drug with respect to both median values and variability [19, 21, 22]. The measurement of enzyme activity (activity per unit amount of CYP as opposed to activity per mg protein provided by liver systems), together with knowledge of the variability of CYP abundances in different populations (with regard to ethnicity, disease, age, etc.), allows variability in in vivo metabolic clearance to be assessed [23]. In this study, parameters were taken from the North European Caucasian population library within Simcyp [20].
We previously developed an efavirenz in IVIVE model for the investigation of DDIs, which was also used for this study [19, 22]. An in vitro-based physiologically based pharmacokinetic model in Simcyp for maraviroc was previously described by Hyland et al. [21]. In vitro data describing nevirapine physiochemical and metabolic characteristics are summarised in Table 1. Data describing the metabolism of nevirapine by different recombinant enzyme isoforms are available in the literature [11, 23]. Induction of CYP3A4 and CYP2B6 are also described [23].
2.2 Simulation Clinical Trial Design
Virtual clinical trials of 100 Caucasian individuals—50 % male with age between 18 and 65 years and reference body weight of 70 kg—were simulated for the different switching scenarios.
2.2.1 Switching from Efavirenz to Maraviroc
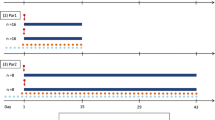
Efavirenz (600 mg once daily) was administered for 21 days followed by maraviroc administration considering four different scenarios (Fig. 2):
-
1.
Maraviroc 600 mg twice daily for 2 weeks followed by standard 300 mg twice-daily dosing.
-
2.
Maraviroc 600 mg twice daily for 1 week followed by standard 300 mg twice-daily dosing.
-
3.
Maraviroc 600 mg twice daily (week 1) followed by 450 mg twice daily (week 2), and standard 300 mg twice-daily dosing thereafter.
-
4.
Maraviroc 450 mg twice daily for 2 weeks followed by standard 300 mg twice-daily dosing.
2.3 Switching from Efavirenz to Nevirapine
Efavirenz (600 mg once daily) was administered for 21 days followed by nevirapine administration considering three different scenarios (Fig. 1):

Schematic representation of the simulated clinical scenarios. a Clinical scenarios switching from efavirenz to maraviroc; b Clinical scenarios switching from efavirenz to nevirapine. BID twice daily, EFV efavirenz, MVC maraviroc, NVP nevirapine, OD once daily
-
1.
Nevirapine 200 mg twice daily after stopping efavirenz.
-
2.
Nevirapine at 200 mg once daily for 2 weeks, after stopping efavirenz, followed by standard 200 mg twice-daily dosing.
-
3.
Two weeks of efavirenz with nevirapine 200 mg once daily, followed by standard 200 mg twice-daily dosing of nevirapine after stopping efavirenz.
In order to assess and validate the models, the results of the simulations were compared with available clinical studies [13, 17, 24].
3 Results
The pharmacokinetics at steady state at a standard dose for the three drugs were initially simulated alone, in order to test the in vitro models and assess the performance of the simulation for each drug (Figs. 2, 3). The results following the switch are summarised in Tables 2 and 3. The simulations predicted pharmacokinetic profiles for each drug consistent with data previously published (Tables 2, 3) [8, 12, 21, 22, 25].

Mean concentrations of maraviroc in plasma with efavirenz (continuous line), and without efavirenz (broken line) at therapeutic dose. Scenario 1: maraviroc 600 mg twice daily for 2 weeks followed by standard 300 mg twice-daily dosing. Scenario 2: maraviroc 600 mg twice daily for 1 week followed by standard 300 mg twice-daily dosing. Scenario 3: maraviroc 600 mg twice daily (week 1) followed by 450 mg twice daily (week 2), and standard 300 mg twice-daily dosing thereafter. Scenario 4: maraviroc 450 mg twice daily for 2 weeks followed by standard 300 mg twice-daily dosing. MVC maraviroc

Mean nevirapine concentrations in plasma with efavirenz (continuous line), and without efavirenz (broken line) at therapeutic dose. Scenario 1: nevirapine 200 mg twice daily after stopping efavirenz (at week 2 nevirapine reached steady state). Scenario 2: nevirapine 200 mg once daily for 2 weeks, after stopping efavirenz, followed by standard 200 mg twice-daily dosing. Scenario 3: 2-week crossover period of efavirenz with nevirapine 200 mg once daily, followed by standard 200 mg twice-daily dosing of nevirapine and stopping efavirenz. NVP nevirapine
3.1 Maraviroc Switch
3.1.1 Scenario 1
After stopping efavirenz therapy, maraviroc was given at an initial dose of 600 mg once daily for 2 weeks, and subsequently reduced to 300 mg twice daily. In this scenario, there was an average decrease of the maraviroc area under the plasma concentration–time curve (AUC) during the initial 2 weeks due to the presence of efavirenz, which is an inducer of CYP3A4. The induction effect of efavirenz was present for more than 25 days after the switch. However, the induction effect was gradually decreasing during this time (Fig. 2).
A previous study by Waters et al. [24] compared the AUC, maximum concentration (C max) and trough concentration (C trough) between day 1 and week 2 (maraviroc 600 mg at steady state) with values at week 4 (maraviroc 300 mg at steady state), reporting a geometric mean ratio of 1.18, 1.48 and 0.59 (day 1 vs. week 4), and 1.99, 1.97 and 2.26 (week 2 vs. week 4), respectively. Our simulations showed a mean ratio of 0.71, 1.08 and 0.56 for day 1 versus week 4, and 1.74, 1.86 and 1.89 for week 2 versus week 4. These results suggested that the in silico model performed adequately, producing simulations within a twofold range of previous clinical trial data, which is normally considered an adequate margin in pharmacokinetic modelling [18, 26].
3.1.2 Scenario 2
The second scenario took into consideration the administration of maraviroc 600 mg twice daily for 1 week followed by 300 mg twice daily. The results were comparable with scenario 1. Scenario 2 showed an average C max at week 1 that was slightly less than the C max at week 2 in scenario 1 (0.833 vs. 0.954 mg/L), which was likely due to the efavirenz effect being more persistent at week 1. Similarly, with the 300 mg regimen there was a slight decrease in C max compared with scenario 1 (15 % less). These differences disappeared after 5 days of therapy.
3.1.3 Scenario 3
An alternative regimen was simulated in scenario 3; a dose of 600 mg in the first week and 450 mg in the second week, followed by the standard dose of 300 mg. This scenario allowed a gradual decrease in the dose over the 2-week period. This switching would potentially avoid high C max values, which are associated with hypotension [10], in particular in the second week of therapy.
3.1.4 Scenario 4
In scenario 4 we considered a dose of 450 mg for the whole 2 weeks followed by the standard dose. This strategy showed a decrease in the plasma concentrations during the initial 2 weeks, but they were still above the suggested concentrations required [C trough and average concentration (C avg) <0.025 and <0.075 mg/L, respectively] for optimal virological response [8, 27] (Fig. 2).
3.2 Efavirenz Pharmacokinetics
Efavirenz intake was stopped on day 30 at steady state, and concentrations were simulated for the following 25 days after stopping. Mean concentrations were above 1 mg/L up to day 7. These results were similar to previously published data [28, 29].
3.3 Nevirapine Switch
3.3.1 Scenario 1
The scenario of immediate full-dose nevirapine, i.e. 200 mg twice daily after stopping efavirenz, was suggested by Winston et al. [17]. The simulated data showed results within twofold of the clinical study (Table 3), which confirmed the model could adequately describe this DDI.
3.3.2 Scenario 2
Scenario 2, 200 mg of nevirapine once daily administered for 2 weeks after stopping efavirenz, followed by standard dosing of nevirapine 200 mg twice daily, was also evaluated by Winston et al. [17]. The results showed a sub-therapeutic concentration of nevirapine during the initial 2 weeks of the switch.
3.3.3 Scenario 3
Dufty et al. [13] suggested a 2-week period of efavirenz with nevirapine 200 mg once daily, followed by standard 200 mg twice-daily dosing of nevirapine after stopping efavirenz. The simulations resulted in a sub-therapeutic concentration of nevirapine during the co-administration. However, the efavirenz C avg values during that period were significantly above the suggested efavirenz minimum effect concentration (1 mg/L). The presence of the two non-nucleoside reverse transcriptase inhibitors should maintain an adequate virological control at the time of the switch (Fig. 3). The simulation results were within twofold of the clinical data [13].
4 Discussion
In clinical practice, antiretroviral regimens are frequently interrupted and modified for various reasons. The most frequently mentioned reasons for changing antiretroviral regimens are intolerance and toxicity. Efavirenz is generally well-tolerated; however, CNS adverse effects are common, can have an impact on adherence and cause therapy interruption [30].
In the present study we used a model simulation based on in vitro data obtained from the literature to predict the human in vivo pharmacokinetics, and to describe the DDIs during the switch from efavirenz to maraviroc or nevirapine. Several switching strategies were simulated. The models described novel strategies and also reproduced previous clinical study data in order to assess the predictive power of the models. This study is an example of the potential use of IVIVE for simulating relevant clinical scenarios.
Waters et al. [24] investigated the pharmacokinetics, safety and efficacy of switching efavirenz to maraviroc in patients taking suppressive efavirenz-based antiretroviral therapy. The study included 11 subjects with undetectable viral load and assessed the pharmacokinetics, efficacy and safety of maraviroc administered at 600 mg twice daily for 2 weeks to HIV-1-infected patients who had achieved viral suppression on efavirenz-based therapy, followed by maraviroc 300 mg twice daily.
Simulations of the switching scenario strategy were carried out using 100 subjects for a time course of 25 days during which they reached steady state (computational limitations constrained simulation further along the time course). The simulated mean maraviroc C trough values were similar to those determined by Waters et al. [24], with both experimentally observed and simulated C trough values exceeding the C trough and C avg values of <0.025 and <0.075 mg/L, respectively (concentrations previously associated with near-maximal virological responses) for the wild-type virus [8, 27]. The simulated C trough values were slightly higher than the clinically observed data (within a twofold window). Both observed and simulated data indicate that the maraviroc C trough at day 6 is similar to the C trough at week 4, suggesting that 1 week could be sufficient to compensate for the prolonged induction effect of efavirenz.
Simulated C max values after 1 week were generally 50 % higher than at week 4 after the switch. A C max near to 1 mg/L after 1 week following the switch could increase the risk of hypotension. Lowering the dose during the initial 2 weeks of the switch from 600 to 450 mg could prevent this risk. In order to test this hypothesis, scenarios 3 and 4 were simulated. These scenarios showed mean C trough values at week 2 similar to mean C trough values in scenario 1 at week 2 with 600 mg of maraviroc. This result suggests the possibility of lowering the dose of maraviroc for the first 2 weeks after the switch, thereby maintaining a therapeutic concentration.
Individuals showing intolerance to efavirenz are often switched to nevirapine. In recent years four main strategies have been proposed. Schouten et al. suggested starting the switch with nevirapine 200 mg once daily for 14 days and then 200 mg twice daily [1]; Winston et al. [17] recommended the full dose of nevirapine without 2 weeks dose escalation; Dufty et al. [13] suggested efavirenz being maintained for 2 weeks with nevirapine 200 mg once daily, then stopping efavirenz and increasing nevirapine to 200 mg twice daily; and Parienti et al. [31] recommended determining efavirenz concentrations before switch to help decide whether to start nevirapine at 200 or 400 mg per day. The present study simulated these scenarios and showed consistency with the clinical results. Interestingly, in all the simulations the efavirenz effect on nevirapine concentrations was negligible. This was consistent with a study by Veldkamp et al. [32], which reported nevirapine concentrations being unaffected by the co-administration of efavirenz when used in combination (Fig. 3).
Based on the simulation results, a dose escalation of nevirapine or commencing at the full dose when switching from efavirenz can be considered. However, the dose escalation could result in a sub-therapeutic concentration of nevirapine for the initial 2 weeks of the switch. In order to have an adequate therapeutic coverage during the 2-week switching time, efavirenz concentrations should exceed the suggested minimum effective concentration (MEC) of >1 mg/L. However, simulation results indicated that the average efavirenz concentration was above the MEC for about 1 week. The presence of nevirapine could also reduce the efavirenz plasma concentrations, as suggested by a previously published report [32].
Commencing at full dose would give an adequate concentration of nevirapine after 3 days. However, starting the switch with the full dose could increase the risk of adverse effects (including skin rash). However, if switching patients to nevirapine they should have an appropriate CD4 cell count, which should decrease the risk of skin rash [33]. Measuring efavirenz concentrations before switch could help decide the strategy used, as suggested by Parienti et al. [31]. Adequate therapeutic concentrations of efavirenz were shown during a 2-week period of both efavirenz and nevirapine 200 mg once daily, when possible sub-therapeutic nevirapine concentrations can occur; this strategy would minimise the risk of rash or hepatotoxicity. Simulation results showed that in the presence of nevirapine at day 14 of the combination, the C max and minimum concentration of efavirenz decreased by 27 and 26 %, respectively.
5 Conclusion
Although the validation of the models was restricted by the presence of limited clinical data, we were able to reproduce some of the already documented DDI clinical studies, observing results which were well within a twofold range of the experimental data.
Broadly, the simulation approach described here provides a means to integrate available information regarding DDIs and to test their impact in different or novel clinical scenarios. In being able to predict changes and variability in pharmacokinetic variables such as C max, C trough and AUC, the approach has the potential to assist in addressing important questions regarding clinical trial study design, such as choosing the appropriate treatment and/or dosing strategy.
This approach has the potential to decrease the possible risk of adverse drug reactions when considering HIV treatment. HIV patients may have to experience several switches of therapies, and applying the optimal strategy can be crucial. Moreover, this approach could be used to consider the characteristics and needs of the individual patient or specific population of patients.
References
Schouten JT, Krambrink A, Ribaudo HJ, Kmack A, Webb N, Shikuma C, et al. Substitution of nevirapine because of Efavirenz Toxicity in AIDS Clinical Trials Group A5095. Clin Infect Dis. 2010;50(5):787–91.
Scourfield A, Zheng J, Chinthapalli S, Waters L, Martin T, Mandalia S, et al. Discontinuation of Atripla as first-line therapy in HIV-1 infected individuals. AIDS. 2012;26(11):1399–401.
Cooper DA, Heera J, Goodrich J, Tawadrous M, Saag M, Dejesus E, et al. Maraviroc versus efavirenz, both in combination with zidovudine-lamivudine, for the treatment of antiretroviral-naive subjects with CCR5-tropic HIV-1 infection. J Infect Dis. 2010;201(6):803–13.
Best BM, Goicoechea M. Efavirenz–still first-line king? Expert Opin Drug Metab Toxicol. 2008;4(7):965–72.
Vrouenraets SM, Wit FW, van Tongeren J, Lange JM. Efavirenz: a review. Expert Opin Pharmacother. 2007;8(6):851–71.
Fellay J, Marzolini C, Decosterd L, Golay KP, Baumann P, Buclin T, et al. Variations of CYP3A activity induced by antiretroviral treatment in HIV-1 infected patients. Eur J Clin Pharmacol. 2005;60(12):865–73.
Kuritzkes D, Kar S, Kirkpatrick P. Fresh from the pipeline—maraviroc. Nat Rev Drug Discov. 2008;7(1):15–6.
Abel S, Back DJ, Vourvahis M. Maraviroc: pharmacokinetics and drug interactions. Antivir Ther. 2009;14(5):607–18.
Trancart S, Charreau I, Marchou B, Bocquentin M, Molina JM, Izopet J, et al. Presence of lamivudine or emtricitabine is associated with reduced emergence of nonnucleoside reverse transcriptase inhibitor mutations in an efavirenz-based intermittent antiretroviral treatment regimen. Antimicrob Agents Chemother. 2012;56(3):1655–7.
Abel S, van der Ryst E, Rosario MC, Ridgway CE, Medhurst CG, Taylor-Worth RJ, et al. Assessment of the pharmacokinetics, safety and tolerability of maraviroc, a novel CCR5 antagonist, in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl 1):5–18.
Erickson DA, Mather G, Trager WF, Levy RH, Keirns JJ. Characterization of the in vitro biotransformation of the HIV-1 reverse transcriptase inhibitor nevirapine by human hepatic cytochromes P-450. Drug Metab Dispos. 1999;27(12):1488–95.
Cooper CL, van Heeswijk RP. Once-daily nevirapine dosing: a pharmacokinetics, efficacy and safety review. HIV Med. 2007;8(1):1–7.
Dufty NE, Barrett S, Taylor S. Switching from efavirenz (EFV) to nevirapine (NVP): a novel pharmacological strategy. 15th Annual conference of the British HIV Association British HIV-AIDS Association (BHIVA); Liverpool, UK; 2009.
Gelinck LB, Burger DM. Switching from efavirenz to nevirapine. Clin Infect Dis. 2010;51(3):365 (author reply 365–6).
Luo R, Piovoso MJ, Martinez-Picado J, Zurakowski R. Optimal antiviral switching to minimize resistance risk in HIV therapy. Plos One. 2011;6(11):e27047.
Ward DJ, Curtin JM. Switch from efavirenz to nevirapine associated with resolution of efavirenz-related neuropsychiatric adverse events and improvement in lipid profiles. AIDS Patient Care STDS. 2006;20(8):542–8.
Winston A, Pozniak A, Smith N, Fletcher C, Mandalia S, Parmar D, et al. Dose escalation or immediate full dose when switching from efavirenz to nevirapine-based highly active antiretroviral therapy in HIV-1-infected individuals? AIDS. 2004;18(3):572–4.
Chen Y, Jin JY, Mukadam S, Malhi V, Kenny JR. Application of IVIVE and PBPK modeling in prospective prediction of clinical pharmacokinetics: strategy and approach during the drug discovery phase with four case studies. Biopharm Drug Dispos. 2012;33(2):85–98.
Siccardi M, Marzolini C, Seden K, Almond L, Kirov A, Khoo S, et al. Prediction of drug-drug interactions between various antidepressants and efavirenz or boosted protease inhibitors using a physiologically based pharmacokinetic modelling approach. Clin Pharmacokinet. 2013;52(7):583–92.
Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The Simcyp population-based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5(2):211–23.
Hyland R, Dickins M, Collins C, Jones H, Jones B. Maraviroc: in vitro assessment of drug-drug interaction potential. Br J Clin Pharmacol. 2008;66(4):498–507.
Siccardi M, Almond L, Schipani A, Csajka C, Marzolini C, Wyen C, et al. Pharmacokinetic and pharmacodynamic analysis of efavirenz dose reduction using an in vitro-in vivo extrapolation model. Clin Pharmacol Ther. 2012;92(4):494–502.
Faucette SR, Zhang TC, Moore R, Sueyoshi T, Omiecinski CJ, LeCluyse EL, et al. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther. 2007;320(1):72–80.
Waters L, Newell S, Else L, Cevik M, Rockwood N, Jackson A, et al. Pharmacokinetics, efficacy and safety of switching from efavirenz to maraviroc twice-daily in patients suppressed on an efavirenz-containing regimen as initial therapy. European AIDS Conference—EACS; 12–15 Oct 2011; Belgrade, Serbia.
Schipani A, Wyen C, Mahungu T, Hendra H, Egan D, Siccardi M, et al. Integration of population pharmacokinetics and pharmacogenetics: an aid to optimal nevirapine dose selection in HIV-infected individuals. J Antimicrob Chemother. 2011;66(6):1332–9.
De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RAHJ. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35(10):1766–80.
Okoli C, Siccardi M, Thomas-William S, Dufty N, Khonyongwa K, Ainsworth J, et al. Once daily maraviroc 300 mg or 150 mg in combination with ritonavir-boosted darunavir 800/100 mg. J Antimicrob Chemother. 2012;67(3):671–4.
Boffito M, Jackson A, Lamorde M, Back D, Watson V, Taylor J, et al. Pharmacokinetics and safety of etravirine administered once or twice daily after 2 weeks treatment with efavirenz in healthy volunteers. J Acquir Immune Defic Syndr. 2009;52(2):222–7.
Sadiq ST, Fredericks S, Khoo SH, Rice P, Holt DW. Efavirenz detectable in plasma 8 weeks after stopping therapy and subsequent development of non-nucleoside reverse transcriptase inhibitor-associated resistance. AIDS. 2005;19(15):1716–7.
Clifford DB, Evans S, Yang Y, Acosta EP, Goodkin K, Tashima K, et al. Impact of efavirenz on neuropsychological performance and symptoms in HIV-infected individuals. Ann Intern Med. 2005;143(10):714–21.
Parienti JJ, Peytavin G, Reliquet V, Verdon R, Coquerel A. Pharmacokinetics of the treatment switch from efavirenz to nevirapine. Clin Infect Dis. 2010;50(11):1547–8.
Veldkamp AI, Harris M, Montaner JSG, Moyle G, Gazzard B, Youle M, et al. The steady-state pharmacokinetics of efavirenz and nevirapine when used in combination in human immunodeficiency virus type 1-infected persons. J Infect Dis. 2001;184(1):37–42.
Manosuthi W, Sungkanuparph S, Tansuphaswadikul S, Inthong Y, Prasithsirikul W, Chottanapund S, et al. Incidence and risk factors of nevirapine-associated skin rashes among HIV-infected patients with CD4 cell counts <250 cells/μL. Int J STD AIDS. 2007;18(11):782–6.
DrugBank. http://www.drugbank.ca. Accessed 22 May 2014.
Cheeseman SH, Hattox SE, McLaughlin MM, Koup RA, Andrews C, Bova CA, et al. Pharmacokinetics of nevirapine: initial single-rising-dose study in humans. Antimicrob Agents Chemother. 1993;37(2):178–82.
Fayet A, Beguin A, de Tejada BM, Colombo S, Cavassini M, Gerber S, et al. Determination of unbound antiretroviral drug concentrations by a modified ultrafiltration method reveals high variability in the free fraction. Ther Drug Monit. 2008;30(4):511–22.
Yazdanian M, Glynn SL, Wright JL, Hawi A. Correlating partitioning and caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm Res. 1998;15(9):1490–4.
Lamson MJ, Sabo JP, MacGregor TR, Pav JW, Rowland L, Hawi A, et al. Single dose pharmacokinetics and bioavailability of nevirapine in healthy volunteers. Biopharm Drug Dispos. 1999;20(6):285–91.
Acknowledgment
We would like to thank the Simcyp team for technical support.
Disclosures
Transparency declarations: David Back and Saye Khoo have received research grants and travel bursaries from Merck, Bristol-Myers Squibb, GlaxoSmithKline, Abbott, ViiV, Boehringer Ingelheim and Janssen. The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Schipani, A., Back, D., Owen, A. et al. Use of In Vitro to In Vivo Extrapolation to Predict the Optimal Strategy for Patients Switching from Efavirenz to Maraviroc or Nevirapine. Clin Pharmacokinet 54, 107–116 (2015). https://doi.org/10.1007/s40262-014-0184-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-014-0184-8