
Abstract
Background and Objectives
Yimitasvir is a novel oral hepatitis C virus non-structural protein 5A (NS5A) inhibitor. The aims of this first-in-human study were to evaluate the safety, tolerability and pharmacokinetics of single and multiple doses of yimitasvir in healthy adult Chinese volunteers and to assess the effect of food on yimitasvir pharmacokinetics.
Methods
Randomized, double-blind, placebo-controlled, single-ascending-dose (30, 100, 200 and 400 mg) and multiple-ascending-dose (100 and 200 mg once daily for 7 days) studies were performed in 32 and 24 subjects, respectively, in male and female adults. Additionally, the effect of food on yimitasvir pharmacokinetics was assessed with a crossover study in 15 male subjects.
Results
Yimitasvir was absorbed slowly after oral administration with a median time to maximum plasma concentration (Tmax) of 3.5–4.0 h. Increases in the maximum plasma concentration (Cmax) and area under the concentration–time curve from 0 to the last measurable time point (AUC0-t) were proportional to the dose of yimitasvir over a dose range of 30–100 mg, while increases were less than dose proportional over a dose range of 200–400 mg in part 1, indicating that absorption at the 200-mg dose was nearly saturated. The geometric mean terminal half-life of yimitasvir was 13.4–19.7 h in each cohort, supporting once-daily dosing. Faecal excretion of parent yimitasvir was the major route of elimination. Steady state was achieved following 5 days of dosing with minimal accumulation. A standardized high-fat meal decreased the rate and extent of absorption. All doses of yimitasvir were well tolerated.
Conclusions
Yimitasvir, at single doses of 30–400 mg and multiple doses of 100–200 mg for 7 days, was well tolerated in healthy Chinese subjects. The results of this study formed the basis for the dosing schemes evaluated in a phase Ib study and subsequent phase II and phase III clinical studies.
Clinical Trial Registration
This study was registered at the China Food and Drug Administration (Registration numbers: 2014L02064 and 2014L02065) and at http://www.chictr.org.cn (Nos. CTR20140854, CTR20150048 and CTR20150123).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Yimitasvir was well tolerated in healthy Chinese subjects up to a 400-mg single dose and 200 mg/day for 7 days. |
The pharmacokinetic profile showed a long half-life, supporting once-daily dosing. |
The exposure of yimitasvir decreased after a standardized high-fat meal. |
1 Introduction
Chronic hepatitis C virus infection is a major public health problem, and over 150 million people worldwide are estimated to be infected with hepatitis C virus (HCV) [1]. It is estimated that at least 25 million individuals are infected with HCV in China [2]. Long-term HCV infection can cause chronic liver diseases, including cirrhosis and hepatocellular carcinoma (HCC).
In recent years, direct-acting antivirals (DAAs), a broad class of potent drugs, have been approved for HCV treatment. Combination regimens of DAAs provide rates of sustained virological response exceeding 90% [3]. However, Peg-interferon-α/ribavirin, which has limited efficacy and significant side effects, is still the current standard treatment in mainland China [2]. There is a crucial need to develop novel, safe and effective DAAs to help patients with HCV.
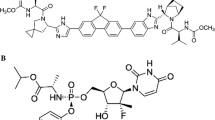
Non-structural protein 5A (NS5A) has been identified to be a target for inhibiting the replication of HCV [4]. Several NS5A inhibitors are used as part of multidrug regimens to treat HCV [5]. Yimitasvir phosphate (Fig. 1) is an NS5A inhibitor, and its structure is similar to daclatasvir [6] and ledipasvir [7]. Preclinical characterization of yimitasvir showed that it has superior or comparable in vitro inhibition efficacy in HCV genotypes 1a, 1b and 4a, which makes yimitasvir a promising new DAA drug for HCV treatment [8].

Chemical structure of yimitasvir phosphate
The objectives of this first-in-human study were to evaluate the safety and tolerability of single- and multiple-dose administration of yimitasvir in healthy adult Chinese volunteers and to assess the pharmacokinetic profile of yimitasvir over the same timeframe. Additionally, the effect of food on pharmacokinetics and tolerability was evaluated.
2 Methods
The protocol and informed consent documentation were reviewed and approved by Peking University First Hospital Ethical Committee (Peking, China), and the study was conducted in accordance with the Declaration of Helsinki and followed the principles of Good Clinical Practice. Written informed consent was obtained for all subjects prior to participation in any study procedures. This phase I study was conducted at Peking University First Hospital Phase I Unit.
2.1 Study Design
The study comprised three parts, as follows (Fig. 2):

Study flow diagram
Part 1 was a single-ascending-dose (SAD) study with four dose cohorts of yimitasvir (30 mg, 100 mg, 200 mg and 400 mg). Eight subjects in each dose cohort were randomized to receive a single oral dose of yimitasvir phosphate capsule (six subjects) or a matching placebo (two subjects) after overnight fasting. Dose escalation was performed after the safety profile of the preceding dose was evaluated.
Part 2 was a multiple-ascending-dose (MAD) study with 100 mg and 200 mg of yimitasvir once daily for 7 days. Twelve subjects in each dose cohort were randomized at a 3:1 ratio to receive multiple doses of yimitasvir phosphate capsule or placebo. The investigators and subjects were blinded to subject randomization throughout the study. Doses were escalated following review of safety in earlier study cohorts.
Subjects in part 1 and part 2 attended a screening visit within 7 days prior to receiving the first dose of study drug. They were admitted to the clinical study unit on day − 1. The day after admission, subjects received their randomized treatment and underwent a period of observation and assessment in the unit (7 days for part 1 and 12 days for part 2). Subjects returned for a follow-up visit on day 10 for part 1 and day 15 for part 2.
Part 3 was a randomized, two-period, two-sequence, open-label, crossover study of yimitasvir with a washout period of 12 days between the two periods. Fifteen subjects were randomized to receive a standard high-fat and high-calorie (at least 800 kcal) breakfast 30 min prior to the administration of a single 100-mg oral dose of yimitasvir or fast prior to drug administration in period 1 or period 2. After dosing, subjects underwent observation and assessment in the unit for 5 days during each period and returned for a follow-up visit on day 8 after the second dosing.
2.2 Study Populations
Healthy Chinese male and female adults (only males in part 3) between 18 and 45 years of age, with a body mass index (BMI) within the range of 19–25 kg/m2 and a weight of at least 50 kg, were eligible to participate in this study. Subjects could participate in only a single cohort and were not allowed to participate in other parts. The key exclusion criteria included: (1) clinically significant abnormalities in the relevant physical examination, vital signs, electrocardiogram (ECG), or clinical laboratory evaluations at screening; (2) positivity for HBV, HCV, HIV or TPPA; (3) use of known inhibitors or inducers of drug metabolism within the previous month or any medication (prescription medication, over-the-counter medication and herbal products) within 2 weeks; (4) history or presence of any systemic disorders or disease; (5) alcohol consumption averaging > 20 g/week within 6 months; (6) regular smoking habit (> 1 cigarettes/day); (7) positive drug screen; (8) participation in an investigational drug/device study within 3 months; (9) blood donation within the previous 3 months or intention to donate blood within 1 month after the trials; or (10) participants or their partners were unwilling to use effective contraception for 3 months after administration of the drug.
2.3 Blood, Urine and Faeces Sampling
To measure yimitasvir plasma levels in part 1 and part 3, a series of blood samples were collected in heparin lithium tubes at predetermined time points: pre-dose and at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, 72, 96, 120 and 144 h (120 and 144 h were not applicable for part 3) post-dose following single-dose administration. In part 2, blood samples were drawn pre-dose and at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12 and 24 h post-dose on day 1 and pre-dose on days 5, 6 and 7 and at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, 72, 96 and 120 h after the last dose. At each point, a 4-ml blood sample was collected and centrifuged at 3000 rpm for 10 min at 4 °C. The plasma samples were stored at − 70 °C.
In part 1, urine samples were obtained pre-dose and at the following predetermined collection intervals: 0–8 h, 8–24 h, 24–48 h, 48–72 h, 72–96 h, 96–120 h and 120–144 h. After measurement of the total volume of urine for each collection timepoint, 10 ml was stored for drug assays. Faeces samples voided 0–144 h post-dose were pooled for the 100- and 200-mg doses in part 1. All of the samples were stored at − 70 °C.
2.4 Pharmacokinetic Assessment
Plasma, urine and faeces samples were prepared using protein precipitation prior to analysis. A Shimadzu Prominence 20-Series liquid chromatography-SCIEX API4000 mass spectrometer was employed to detect yimitasvir and d8-DAG (isotope-labelled yimitasvir as an internal standard). The analytical column was a Waters Xterra MS C18 (50 × 2.1 mm; 5 μm; Waters Technologies, Inc., Milford, MA, USA). Mobile phase A was 0.1% formic acid and 5 mM ammonium formate in water, and mobile phase B was 0.1% formic acid acetonitrile for plasma, urine and faeces samples. The injection volume was 2 μl, and the flow rate was 0.4 ml/min for plasma and urine samples. The injection volume was 5 μl, and the flow rate was 0.4 ml/min for faeces samples. The column temperature was maintained at 40 °C, and the mass spectrometer was used in the positive scan mode. Quantitation was accomplished by triple quadrupole mass spectrometry and monitoring of the precursor-to-production pairs m/z 856.5–630.2 for yimitasvir and m/z 864.4–630.5 for the internal standard.
The linear calibration ranges were 5.00–5000 ng/ml for plasma, 1.00–900 ng/ml for urine and 100–100,000 ng/ml for faeces. The lower limits of quantitation (LLOQs) were 5.00 ng/ml for plasma, 1.00 ng/ml for urine, and 100 ng/ml for faeces. The four QC sample concentrations were 5.00, 15.0, 250 and 4000 ng/ml for plasma; 1.00, 3.00, 50.0 and 800 ng/ml for urine; and 100, 300, 5000 and 80,000 ng/ml for faeces. The inter- and intra-day precision values (RSD %) were < 11.7%, and accuracy was within 92.8–107.8% for plasma; the inter- and intra-day precision values (RSD %) were < 9.7%, and accuracy was within 97.7–104% for urine; and the inter- and intra-day precision values (RSD %) were < 7.9%, and accuracy was within 92–103% for faeces. No significant matrix effect was found. All samples were analysed within established storage stability periods. Yimitasvir in plasma was stable on the bench-top for 24 h, six freeze/thaw cycles, and 167 days at − 10 °C to − 30 °C and − 60 °C to − 80 °C. Yimitasvir in urine was stable on the bench-top for 25 h, five freeze/thaw cycles, and 222 days at − 10 °C to − 30 °C and − 60 °C to − 80 °C. Yimitasvir in faeces was stable on the bench-top for 24 h, five freeze/thaw cycles, and 218 days at − 10 °C to − 30 °C and − 60 °C to − 80 °C.
Pharmacokinetic analyses were performed to determine the plasma pharmacokinetic parameters and the total excretion of yimitasvir in urine and faeces by standard noncompartmental methods using Phoenix WinNonlin 6.3 (A Certara™ Company, Princeton, NJ, USA). Maximum concentration (Cmax), Cmax at steady state after multiple doses (Cmax,ss), minimum concentration at steady state after multiple doses (Cmin,ss), and the time to Cmax or Cmax,ss (Tmax and Tmax,ss) were obtained from the observed data. The area under the curve (AUC) for plasma concentration versus time from 0 to the last measurable time point (AUC0−t), the AUC from 0 to infinity (AUC0−∞), the AUC over a dosing interval at steady state (AUCtau), the terminal half-life (t1/2), the apparent plasma clearance after extravascular administration (CL/F), the apparent volume of distribution (Vz/F) and the accumulation ratio (Racc,observed = day 7 AUC0–24/day 1 AUC0–24, Racc,predicted = 1/(1 − e−λz×24)) were calculated.
In addition, accumulative urine and faeces excretion was calculated as the sum of the amounts excreted during each interval and expressed as accumulative excretion amount (Au0–t) and accumulative excretion ratio (Fe0–t).
2.5 Safety Assessment
Physical examinations, electrocardiograms (ECGs), assessment of vital signs and standard clinical laboratory evaluations (haematology, blood chemistry, lymphocyte subpopulations, blood coagulation function and urinalysis) were performed at screening and periodically after dosing. Adverse events (AEs) and the concomitant medications taken were recorded from time of first administration of the study drug until study completion. All AEs were evaluated by the investigators for severity and relationship to the study drug.
2.6 Statistical Methods
Descriptive statistics were performed for AEs, vital signs, ECG results, and clinical laboratory abnormalities. Dose proportionality was assessed across doses for Cmax and AUC values using Power Model [9, 10]. The food effect on yimitasvir pharmacokinetics was assessed in part 3 using analysis of variance (ANOVA). The 90% confidence interval (CI) of the ratio of geometric means for the variables AUC0–t, AUC0–∞ and Cmax were calculated for the fed and fasted conditions. Tmax was evaluated by a nonparametric test. Statistical analyses were performed using SAS 9.3 software (SAS Institute Inc, Cary, NC, USA).
3 Results
3.1 Demographic Characters
In part 1, 32 healthy native Chinese subjects were randomized and completed the study (n = 24 for yimitasvir and n = 8 for placebo). Twenty-four healthy subjects (n = 18 for yimitasvir and n = 6 for placebo) were enrolled in part 2, and one subject discontinued the study because of an AE, urticaria. In part 3, 15 subjects were enrolled, and 14 completed the study. One subject was lost to follow-up before period 2. All subjects with evaluable data were included in the safety analysis. Twenty-four, 18 and 14 subjects, respectively, were included in the pharmacokinetic analysis in parts 1, 2 and 3. Demographic data are summarized in Table 1.
3.2 Pharmacokinetic Assessments
The arithmetic mean plasma concentration versus time profiles are presented in Figs. 3, 4 and 5. The principal pharmacokinetic parameters for the three study parts are summarized in Tables 2, 3 and 4. In part 1, the median Tmax of the four dose cohorts ranged from 3.5 h to 4.0 h. The geometric mean t1/2 was 13.4–19.7 h over the dosage range. The geometric mean Cl/F and VZ/F showed increasing trends as the dose increased. Based on the power model assessment (Fig. 6), there was a significant deviation from dose proportionality for Cmax, AUC0–t and AUC0–∞ (slope 0.681, 90% CI (0.500–0.861); slope 0.732, 90% CI (0.571–0.892); slope 0.712, 90% CI (0.556–0.868), respectively). Increases in the Cmax and AUC0–t were proportional for the doses ranging from 30 mg to 100 mg. However, over the dose range of 200–400 mg, the increase in the AUC and Cmax was less than dose proportional. No significant difference in pharmacokinetic parameters was observed between male and female subjects.

Mean plasma concentration–time profile of yimitasvir in part 1. a Linear scale; b semi-log scale. Error bars represent the standard deviations

Mean plasma concentration–time profile of yimitasvir in part 2. a Linear scale; b semi-log scale. Error bars represent the standard deviations

Mean plasma concentration–time profile of yimitasvir in part 3. a Linear scale; b semi-log scale. Error bars represent the standard deviations

Dose proportionality assessment. aCmax, b AUC0–t and c AUC0–∞ versus dose of yimitasvir. Cmax maximum concentration, AUC0–t area under the concentration–time curve from 0 to the last measurable time point, AUC0–∞ area under the concentration–time curve from 0 to infinity
In part 2, plasma concentrations reached steady state by day 5 following repeated dose administration. The plasma concentration–time profile on day 1 was similar to that on day 7. The Cmax and AUC0–t on day 1 were 472 and 689 ng/ml and 5720 and 7650 ng·h/ml for 100 mg and 200 mg, respectively. The corresponding values on day 7 were 635 and 1050 ng/ml and 11,500 and 17,000 ng h/ml, respectively. The predicted accumulation ratios were 1.32 and 1.34 for 100 mg and 200 mg, respectively. The observed accumulation ratios were 1.42 and 1.59, respectively, indicating no significant accumulation of yimitasvir. The accumulation ratio of daily dosing of yimitasvir was in agreement with the t1/2 of yimitasvir administered on a once-daily regimen. Values of Cmax and AUC0-t increased in a less-than-dose-proportional manner from 100 to 200 mg.
In part 3, after a standard high-fat breakfast, the extent of absorption was affected significantly by food intake, as the ratio of geometric means (90% CI) in fed versus fasting conditions was 37.0% (27.6%, 49.4%) for Cmax, 52.3% (39.5%, 69.4%) for AUC0–t, and 53.8% (41.1%, 70.6%) for AUC0–∞. Tmax was significantly delayed from a median of 4.0 h under the fasting condition to 10.0 h under the fed condition (P = 0.00190).
Urinary and faecal excretion of yimitasvir was evaluated in single-dose cohorts for 100 mg and 200 mg. Table 5 presents the accumulative excretion amount and accumulative excretion ratio of yimitasvir. Less than 0.04% of yimitasvir 100 mg and 200 mg was excreted in urine as the parent drug. However, approximately 75.9% and 62.0% of the drug in the 100 mg and 200 mg doses, respectively, was excreted as the parent drug in faeces.
3.3 Safety and Tolerability
The safety analysis included data from all 71 subjects who received at least one dose of yimitasvir (n = 57) or placebo (n = 14). Summaries of the AEs reported during the study are provided in Table 6.
In part 1, 12 of the 24 subjects administered yimitasvir developed 21 AEs, and of these, only four AEs were related to the drug. No AEs were reported in subjects administered placebo. In part 2, the incidence of AEs was similar in yimitasvir (27.8%) and placebo (33.3%) subjects. In part 3, two subjects reported two AEs in the fasted period and one subject reported two AEs in the fed period. One subject was lost to follow-up before period 2 dosing.
The most frequently reported AEs were low potassium and elevated creatine kinase levels. Both occurred in four of 57 subjects who were exposed to yimitasvir. However, these two AEs were recognized as possibly unrelated to the study drug. Elevated alanine transaminase (ALT), aspartate transaminase (AST) and bilirubin (TBIL) levels were observed in our studies and were considered by the investigator to be related to yimitasvir. However, these parameters did not increase in a dose-dependent manner. No clinically meaningful change was observed in physical examination results, vital sign measurements or ECG.
Yimitasvir was well tolerated without serious adverse events (SAEs) or dose-limited toxicity. All AEs were mild in severity, except moderate urticaria, which occurred in one subject who received placebo and withdrew from the part 2 study, and which resolved after antiallergic treatment. No dose-related trends with regard to incidence or intensity of adverse events were observed.
4 Discussion
In this first-in-human study, the safety, tolerability and pharmacokinetics of yimitasvir were evaluated in healthy Chinese human volunteers by administration of single and multiple once-daily ascending doses as well as under fed and fasting conditions. Both single and multiple doses of yimitasvir were well tolerated at the 30-, 100-, 200- and 400-mg dose levels given after overnight fasting. There were no deaths or serious AEs; all observed AEs were mild to moderate in severity. No obvious dose-related trends in the frequency of specific adverse events were observed, and similar findings were obtained to those of the phase I studies of Velpatasvir, Pibrentasvir and GSK2336805 [11,12,13].
Following single- or multiple-oral-dose administration, yimitasvir was absorbed slowly, with peak exposure being reached at 3.5–4.0 h. Dose-proportional linear kinetics were observed over the dose range of 30–100 mg, whereas the increases in plasma exposure (Cmax and AUC0–t) between 200 mg and 400 mg in part 1 and between 100 mg and 200 mg in part 2 were less than the dose increase. It was concluded that 200 mg may have a tendency to saturate absorption and that 400 mg may be close to the maximum saturated dose. We suspected that this finding was owing to the poor solubility of this drug in the intestine, where drug precipitated in intestinal fluid and affected the absorption. Compared with the fasted state, food decreased the extent and rate of absorption of yimitasvir, shown by a decrease of 63.0% in Cmax and 47.7% in AUC0–72 h and a delay in median Tmax from 4 to 10 h. Yimitasvir is a weak base with pH-dependent solubility. In vitro solubility data demonstrated that the solubility decreased along with an increase in pH from 2 to 6.8. In the fasting condition, yimitasvir solubility increased due to a low gastric pH in the stomach. However, a high-fat meal containing protein, fat and carbohydrate resulted in higher gastric pH and fluid volume available for drug dissolution reduction [14]; both could have a negative effect on the yimitasvir dissolution and absorption extent. The decrease in absorption rate might be attributed to food, which can lengthen gastric emptying time and reduce the contact area between yimitasvir and the gastrointestinal tract [15]. These results in healthy volunteers clearly suggest that yimitasvir should be given under fasting conditions.
In this study, a total of three metabolites were detected in human plasma samples, and all were mono-oxide metabolites at low proportions (data not shown). Therefore, yimitasvir was mainly eliminated as the parent drug. A mass balance study indicated that yimitasvir was excreted mainly in faeces, and very little was excreted through the urine as the parent drug. After reaching the peak concentration following single administration, yimitasvir was eliminated via first-order kinetics. The geometric mean t1/2 was 13.4–19.7 h, which indicated that yimitasvir has low or moderate clearance. The pharmacokinetic parameters on day 1 were similar to those on day 7, indicating that the clearance rate of yimitasvir remained unchanged before and after steady state.
We used the preclinical, clinical efficacy and pharmacokinetic data of daclatasvir [16, 17] to estimate the therapeutic dose of yimitasvir. The clinical dose of daclatasvir was 60 mg. The plasma protein binding rate and liver/blood ratio of yimitasvir were 83.6 and 9–44, respectively (data not published), which were similiar to daclatasvir [18], so the therapeutic dose of yimitasvir could be evaluated as presuming that the ratio of the concentration at 24 h after dose (C24 h) and protein-binding-adjusted EC90 (PBA EC90) were the same as that of 60 mg daclatasvir. The C24 h of daclatasvir was approximately 201 ng/ml at this dose based on its pharmacokinetic parameters, and the PBA EC90 (40% normal human serum adjusted) of daclatasvir was 0.051 ng/ml calculated by GraphPad Prism. The C24 h was 3941-fold higher than the PBA EC90 for daclatasvir (201 ng/ml/0.051 ng/ml = 3941). The PBA EC90 (40% normal human serum adjusted) of yimitasvir was 0.045 ng/ml calculated by GraphPad Prism. According to this value, the optimal C24 h of yimitasvir was calculated as 177 ng/ml (3941 × 0.045 ng/ml). Based on this trial, it was predicted that the optimal dose should be approximately 100 mg/day. This recommended dosage is consistent with the result of a phase Ib study [8]. The estimated geometric mean terminal t1/2 values after multiple dosing (16.4 h for 100 mg and 15.3 h for 200 mg) support a once-daily regimen, which was a regimen that results in good patient compliance compared with more frequent dosing regimens or other routes of administration.
5 Conclusions
In conclusion, this study assessed the safety, tolerability and pharmacokinetics of yimitasvir in healthy adult Chinese volunteers in the presence and absence of food. Yimitasvir was well tolerated in all parts of this trial. The results of this study formed the basis for the dosing schemes evaluated in a phase Ib study and subsequent phase II and phase III clinical studies.
References
Bukh J. The history of hepatitis C virus (HCV): basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J Hepatol. 2016;65(1 Suppl):S2–21.
Bian DD, Zhou HY, Liu S, et al. Current treatment status and barriers for patients with chronic HCV infection in mainland China: a national multicenter cross-sectional survey in 56 hospitals. Medicine (Baltimore). 2017;96(34):e7885.
Bourlière M, Gordon SC, Flamm SL, et al. Sofosbuvir, velpatasvir, and voxilaprevir for previousely treated HCV infection. N Engl J Med. 2017;376:2134–46.
Pietchmann T, Lohmann V, Rutter G, et al. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J Virol. 2001;75:1252–64.
Zeuzem S. Treatment options in hepatitis C. Dtsch Arztebl Int. 2017;114:11–21.
Fridell RA, Qiu D, Wang C, Valera L, Gao M. Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob Agents Chemother. 2010;54:3641–50.
Link JO, Taylor JG, Xu L, et al. Discovery of ledipasvir (GS-5885): a potent, once-daily oral NS5A inhibitor for the treatment of hepatitis C virus infection. J Med Chem. 2014;57:2033–46.
Zhang H, Zhu XX, Li Q, et al. Clinical evaluation of efficacy, tolerability and pharmacokinetics of yimistavir phosphate in patients infected with hepatitis C virus. J Pharm Pharmacol. 2018;70:855–64.
Sheng Y, He Y, Huang X, Yang J, Wang K, Zheng Q. Systematic evaluation of dose proportionality studies in clinical pharmacokinetics. Curr Drug Metab. 2010;11(6):526–37.
Hummel Jürgen, McKendrick Sue, Brindley Charlie, French Raymond. Exploratory assessment of dose proportionality: review of current approaches and proposal for a practical criterion. Pharm Stat. 2009;8(1):38–49.
Mogalian E, German P, Kearney BP, et al. Preclinical pharmacokinetics and first-in-human pharmacokinetics, safety, and tolerability of velpatasvir, a pangenotypic hepatitis C virus NS5A inhibitor, in healthy subjects. Antimicrob Agents Chemother. 2017;61(5):1–12.
Lin CW, Dutta S, Asatryan A, et al. Pharmacokinetics, safety, and tolerability following single and multiple doses of pibrentasvir in a first-in-human study. Clin Pharmacol Drug Dev. 2018;7(1):44–52.
Wilfret DA, Walker J, Adkison KK, et al. Safety, tolerability, pharmacokinetics, and antiviral activity of GSK2336805, an inhibitor of hepatitis C virus (HCV) NS5A, in healthy subjects and subjects chronically infected with HCV genotype 1. Antimicrob Agents Chemother. 2013;57(10):5037–44.
Carver Peggy L, Fleisher David, Zhou Simon Y, Kaul Daniel, Kazanjian Power, Li Cheng. Meal composition effects on the oral bioavailability of indinavir in HIV-infected patients. Pharm Res. 1999;16(5):718–24.
Winstanley PA, Orme ML. The effects of food on drug bioavailability. Br J Clin Pharmacol. 1989;28(9):621–8.
Gao M, Nettles RE, Belema M, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100.
Nettles RE, Gao M, Bifano M, et al. Multiple ascending dose study of BMS-790052, a nonstructural protein 5A replication complex inhibitor, in patients infected with hepatitis C virus genotype 1. Hepatology. 2011;54:1956–65.
The U.S. food & drug administration pharmacology review: daclatasvir (DAKLINZA). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206843Orig1s000TOC.cfm. Accessed 10 May 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by National Major Scientific and Technological Special Project for “Significant New Drugs Development” during the Thirteenth Five-year Plan Period (No. 2017ZX09201006-004) and State Key Laboratory of Anti-Infective Drug Development (Sunshine Lake Pharma Co., Ltd), (No. 2015DQ780357).
Conflict of interest
Yingjun Zhang, Lin Luo, Zhangma Huang, Jing Li, Xingan Wang, Huan Yan, Bixia He, Hongming Xie and Qingyun Ren are employees of Sunshine Lake Pharma Co., Ltd.
Ethical approval
All procedures performed in the trials involving human participants were in accordance with the Declaration of Helsinki and followed the principles of Good Clinical Practice. The protocol and informed consent documentation were reviewed and approved by Peking University First Hospital Ethical Committee (Peking, China).
Informed consent
Informed consent was obtained from all individual subjects included in the trial.
Rights and permissions
About this article

Cite this article
Zhao, N., Xie, R., Zhao, X. et al. Safety, Tolerability and Pharmacokinetics of Yimitasvir Phosphate Capsule, a Novel Oral Hepatitis C Virus NS5A Inhibitor, in Healthy Chinese Volunteers. Clin Drug Investig 39, 671–681 (2019). https://doi.org/10.1007/s40261-019-00791-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-019-00791-8