
Abstract
Background and Objective
Tylerdipine hydrochloride (KBP-5660) is a novel L/T-type dual calcium channel blocker developed for the treatment of hypertension. We aimed to study the pharmacokinetics, safety and tolerability of tylerdipine in healthy Chinese subjects.
Methods
Two double-blind, randomized, dose-escalation studies were conducted that included a total of 88 healthy subjects: (1) a single-ascending dose (SAD) study; and (2) a multiple-ascending dose (MAD) study. In the SAD study, 64 subjects were randomly assigned to receive a single dose of 0.5, 2.5, 5, 10, 15, 20, 25, or 30 mg of tylerdipine or placebo. In the MAD study, 24 subjects were randomly assigned to receive 10 or 20 mg of tylerdipine or placebo once daily for 9 days. Blood samples were collected at the designated time points for pharmacokinetic analyses. Safety assessments were conducted throughout the study.
Results
Following a single oral dose of tylerdipine of 5–30 mg, the mean maximum plasma concentration (Cmax) increased from 0.9993 to 10.11 ng/ml; mean area under the plasma-concentration curve (AUC) from time zero to 72 h increased from 4.332 to 73.95 h·ng/ml. AUC increased in a greater than dose-proportional manner, whereas Cmax exhibited a rough but non-typical dose-proportionality increase. In the MAD study, steady-state conditions were achieved after 1 week of daily dosing in both dose groups. Accumulation of tylerdipine was low, with accumulation ratios (RAUC) of less than 1.65. All adverse events were assessed as mild or moderate.
Conclusion
Tylerdipine hydrochloride was safe and well tolerated. The exposure (AUC) of tylerdipine over the dose range of 5–30 mg increased in a greater than dose-proportional manner, while Cmax exhibited a rough but non-typical dose proportionality increase. A slight accumulation of tylerdipine was observed following multiple dosing.
Study registrations
CTR20140862 and CTR20150660.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Tylerdipine hydrochloride was safe and well tolerated in healthy Chinese subjects. |
The exposure (AUC) of tylerdipine over the dose range of 5–30 mg increased in a greater than dose-proportional manner, while the Cmax exhibited an approximately dose proportionality increase. |
Tylerdipine hydrochloride exhibited a slight accumulation following multiple dosing in healthy Chinese subjects. |
1 Introduction
Hypertension is a common medical and social problem leading to cardiovascular disease, stroke, and kidney disease [1]. The prevalence of hypertension is very high, ranging from 15 to 37% of the total population in different areas of the world [2]. It also has been estimated that 1.56 billion people will suffer from hypertension by 2025 [3]. Reducing blood pressure using antihypertensive drugs is an effective way to decrease the morbidity and mortality induced by hypertension, as well as its complications [4].
The most representative antihypertensive drugs are classified as: angiotensin-converting enzyme inhibitors (ACE inhibitors), beta-adrenergic blockers (β-blockers), calcium (Ca2+) channel blockers (CCBs), and diuretics [5]. Since 2014, CCBs, especially dihydropyridine (DHP) CCBs, have been officially recommended by the Eighth Joint National Committee (JNC 8) as initial monotherapy for hypertension treatment. DHP CCBs, which were introduced in the 1960 s, have undergone several changes to optimize their efficacy and safety [6].
Conventional DHP CCBs, including nifedipine and amlodipine, exert their clinical effects predominantly by blocking L-type Ca2+ channels, and they are more likely to be associated with peripheral edema, headache, tachycardia, facial flush, and reflex tachycardia. Although these adverse effects are not considered life threatening, they can contribute to poor patient compliance, particularly in the cases of peripheral edema [7]. Clinical use of these drugs is significantly limited due to the aforementioned reactions.
Some novel DHP CCBs, such as efonidipine [8], benidipine [9], and nilvadipine [10], have recently been developed and demonstrated to possess blocking activity on T-type, as well as L-type Ca2+ channels in certain specific tissues or organs. In randomized trials, efonidipine and benidipine have been demonstrated to exert similar or superior efficacy to L-type CCBs, with lower rates of peripheral edema and negligible reflex tachycardia [9, 11]. Efonidipine has also been reported to exert vasodilator activity on both afferent and efferent arterioles, subsequently reducing proteinuria and retarding the subsequent progression of chronic kidney disease (CKD) [12, 13]. Accordingly, dual blockade of L- and T-type Ca2+ channels may offer beneficial effects on high blood pressure, confer better cardiovascular or renal protection, and reduce edema formation compared with blockade of L-type Ca2+ channels alone [14]. This factor may account for the differentiated pattern of expression of T-type versus L-type Ca2+ channels in arterioles and venules.
Tylerdipine hydrochloride (KBP-5660) is a novel dual L- and T-type CCB developed by Xuan Zhu Pharma Co., Ltd. for the treatment of hypertension. The chemical structures are shown in Fig. 1 (patent number: CN 201110109496.2). Tylerdipine is primarily metabolized by carboxylesterase and cytochrome P450 (CYP) isoenzymes 3A [15], and both M2 and M4 are the main metabolites of tylerdipine and exhibit no antihypertensive effects. In vitro studies have demonstrated that tylerdipine exhibits potent inhibition on both L- and T-type Ca2+ channels. The compound has shown superior antihypertensive effects to barnidipine and lercanidipine, and similar effects to amlodipine in spontaneously hypertensive rats (SHR) and renovascular hypertensive dogs (data not published). Additionally, significant reductions in heart rate and decreases in proteinuria were observed after administration of tylerdipine to deoxycorticosterone acetate (DOCA)-salt rats. These pre-clinical results support the hypothesis that tylerdipine may be a better antihypertensive agent with organ-protective effects and a lower incidence of peripheral edema. However, the pharmacokinetic profile of tylerdipine after single or multiple oral administration has yet to be reported in humans.

Chemical structures of tylerdipine hydrochloride and its metabolites, M2 and M4
Therefore, the present study was conducted to investigate the safety, tolerability, and pharmacokinetics of tylerdipine after oral administration of single ascending doses of 0.5–30 mg and multiple doses of 10 or 20 mg every 24 h for 9 days in healthy Chinese subjects. These results will facilitate the understanding of the pharmacokinetic properties of tylerdipine and provide a scientific basis for the design of further clinical trials.
2 Subjects and Methods
2.1 Chemicals and Materials
Reference standards for tylerdipine (purity 99.6%), M2 (purity 89.9%), M4 (purity 96.5%), D5-tylerdipine (purity 93.0%, IS1) and D6-M4 (purity 99.4%, IS2) were all provided by Xuan Zhu Pharma Co., Ltd (Jinan, China). HPLC-grade acetonitrile and formic acid were purchased from Merck (Darmstadt, Germany) and ROE Scientific Inc. (Newmark, USA), respectively. Deionized water was obtained by a Millipore water purification system (Millipore, Milford, MA). All other reagents were of reagent grade or better and obtained from commercial sources.
The test drug (tylerdipine hydrochloride) and corresponding placebo were supplied by Xuan Zhu Pharma Co., Ltd. (Jinan, China). Subjects were given the required doses of test drug or placebo for each different treatment.
2.2 Subjects
This study was conducted in compliance with the ethical principles set forth in the Declaration of Helsinki (1989) as well as with local applicable laws and regulations. The study protocol and informed consent were approved by the Ethics Committee of the First Affiliated Hospital with Nanjing Medical University (Nanjing, China). All subjects provided written informed consent prior to enrollment in the study.
Male and female healthy Chinese subjects aged 18–45 years with a body mass index (BMI) of 18–25 kg/m2 and weight ≥ 50 kg were enrolled in this study. They were judged to be in good health according to a medical history, physical examination, vital signs measurement, electrocardiogram (ECG), and standard clinical laboratory tests. Meanwhile, female subjects were required to have a negative serum pregnancy test and be using appropriate contraception throughout the study. Exclusion criteria included: history of drug allergy; systemic disease; low blood pressure with systolic blood pressure (SBP) ≤ 95 mmHg and diastolic BP (DBP) ≤ 65 mmHg; participation in another clinical trial or blood donation (> 400 ml) within 3 months of study enrollment; administration of prescription or herbal products within 2 weeks before the study; and history of alcohol or tobacco abuse.
2.3 Study Design
Two double-blind, randomized, dose-escalation, placebo-controlled studies were conducted to investigate the pharmacokinetics, safety, and tolerability of tylerdipine after administration of single and multiple doses in healthy Chinese subjects.
2.3.1 Single-Ascending Dose (SAD) Study
In this study, 64 healthy subjects were randomly assigned to eight groups to receive single doses of 0.5, 2.5, 5, 10, 15, 20, 25, or 30 mg of tylerdipine or matching placebo. Details were as follows: No placebo treatment was set in the 0.5- and 30-mg dose groups, which comprised four subjects. The 2.5- and 25-mg dose groups comprised eight subjects, two of whom received the placebo. Other dose groups comprised ten subjects, two of whom received placebo. They were admitted to the Phase I Clinical Unit before the study and fasted for at least 10 h before drug administration, subsequently receiving a single oral dose at 8:00 a.m. with 250 ml of water. Drinking water was not allowed for 2 h before or after dosing. A standard lunch was provided 4 h after morning dosing, and an evening meal was served 10 h after dosing. Dose escalation was based on the available safety and tolerability results in the preceding group.
For determining concentrations of tylerdipine and its metabolites (M2 and M4) in plasma, blood samples (4 ml) were collected into tubes, which were anticoagulated with K2EDTA before each dose (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 8, 12, 24, 48, 72, 96, and 120 h after administration. Samples were centrifuged at 4,000 rpm for 5 min, and plasma samples were separated and stored at -80 °C until analysis.
2.3.2 Multiple-Ascending Dose (MAD) Study
Tenty-four healthy Chinese subjects were enrolled in the multiple-ascending dose study. They were orally administered 10 or 20 mg of tylerdipine or placebo once daily for 9 consecutive days; each group comprised 12 subjects, two of whom received the placebo. Intake of water or food was permitted in the same way as that in the SAD study.
Serial blood samples (4 ml) were collected at 0 (pre-dose), 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 8, 12, and 24 h on Day 1. On Days 6, 7, 8, and 9, blood samples were collected before drug administration to determine the trough concentration (Cmin). On the last day (Day 9), blood samples were collected at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 8, 12, 24, 48, 72, 96, and 120 h after drug administration. Plasma was separated and stored the same way as that in the SAD study.
2.4 Pharmacokinetic Assessments
2.4.1 Determination of Tylerdipine and Its Metabolites (M2 and M4)
Plasma drug concentrations of tylerdipine and its metabolites (M2 and M4) at each time point were determined using two different validated LC-MS/MS assay methods.
Briefly, for determination of tylerdipine and its metabolites (M2 and M4) in plasma, plasma samples (100 μl) were spiked with 100 μl of IS (IS1/IS2: 0.5/10 ng·ml−1), and 300 μl of acetonitrile, followed by vortex mixing for 5 min and centrifugation at 16,000 rpm at 4 °C for 10 min. 400 µL of supernatant was transferred to another tube and evaporated to dryness. Dry extracts were re-dissolved in 200 μl of 50% acetonitrile, a 5-μl aliquot of which was injected to determine the concentration of tylerdipine. Meanwhile, 80 μl was spiked with an equal volume of deionized water, mixing by vortex for 30 s, followed by 5 μl of the resulting solution being injected into the liquid chromatography-tandem mass spectrometry (LC-MS/MS) system to determine the concentration of the metabolites M2 and M4.
Compounds were chromatographed on an Agilent Poroshell 120 column (2.7 µm, 50 × 2.1 mm) (Agilent, CA, USA) with acetonitrile: 2 mM ammonium formate (0.1% formic acid) (55:45, v/v for KBP-5660; 25:75, v/v for M2 and M4) at a flow rate of 0.3 ml/min.
Detection was performed by MS/MS using an AB QTRAP5500 mass spectrometer (Applied Biosystems/Sciex, Foster City, CA, USA) in MRM mode. Tylerdipine and IS1 were operated in positive ionization mode, with the strongest transitions at m/z 610.3/278.3 and m/z 615.2/283.3, respectively, while M2, M4, and IS2 were operated in negative ionization mode with the strongest transitions at m/z 329.0/252.8, m/z 331.0/207.7, and m/z 337.0/213.9, respectively.
Methods were validated in terms of specificity, calibration curve, matrix effect, accuracy, precision, stability, and dilution effect. Linear calibration curves were obtained in the concentration range of 0.02–20 ng/ml for tylerdipine, and 0.25–250 ng/ml for its metabolites (M2 and M4), respectively.
2.4.2 Pharmacokinetic Analysis
Pharmacokinetic parameters were estimated by non-compartmental methods with Phoenix WinNonlin (version 6.4, Certara, L.P., Princeton, NJ, USA) using the actual recorded sampling times. Parameters are presented as follows: peak plasma concentration after administration (Cmax), time to reach Cmax (Tmax), area under the concentration-time curve from zero to 24 h (AUC0–24 h) or t (AUC0–t, where t is the time of last measurable sample), AUC from zero to infinity (AUC0–∞), elimination half-life (T1/2), effective half-life (T1/2, eff) [16], apparent clearance (CL/F), and apparent volume of distribution during the terminal phase (Vz/F). Cmax and Tmax were obtained directly from the observed plasma concentration-time values. AUC0–t or AUC0–24 h was calculated using the linear up log down rule. AUC0–∞ was calculated as AUC0–t + Ct/λz, where Ct is the last detected concentration and λz is the slope of the log-linear regression of the terminal declining phase. T1/2 was calculated as ln2/λz using the best fit mode. T1/2, eff was estimated based on drug accumulation at steady state and calculated according to the following equation:\(T_{{1/2, {\text{eff}}}} = - \frac{\tau \times \ln 2}{{\ln \left( {1 - \frac{1}{\text{Ra}}} \right)}}\), where τ was the dosing interval and Ra was the AUC accumulation ratio at steady state [16]. CL/F and Vz/F were estimated as Dose/AUC0–∞ and CL/λz, respectively. Accumulation ratio (Racc) was calculated as AUC0–24 h (Day 9)/AUC0–24 h (Day 1) or Cmax (Day 9)/Cmax (Day 1), respectively.
2.5 Tolerability Assessments
Safety and tolerability assessments included monitoring and recording all adverse events (AEs) and serious AEs. Additional safety assessments included monitoring of vital signs, 12-lead electrocardiograms (ECGs), as well as study of blood chemistry, urinalysis, and hematology. All subjects remained in the study unit and were continuously observed throughout the study. Physicians evaluated all the adverse events according to the intensity (mild, moderate, or serious), duration, outcome, or potential relationship to the test drug. All the information after administration was summarized descriptively for each treatment group.
2.6 Statistical Analysis
All the pharmacokinetic parameters are expressed with the coefficient of variation, with any geometric means indicated, or as the median and range of values. Descriptive statistics are provided for pharmacokinetic concentrations and derived pharmacokinetic parameters. Dose proportionality of tylerdipine in the dose range of 5–30 mg was assessed using the power model [17]: Ln(pharmacokinetic parameters) = β0+ β1Ln(Dose), where pharmacokinetic parameters were the AUC or Cmax, β0 was the intercept, and β1 was the dose proportionality coefficient. The 90% confidence interval (CI) for the β1 was calculated. Furthermore, the predefined criterion intervals for AUC and Cmax were 0.80–1.25 and 0.70–1.43, respectively. According to the doses and predefined criterion intervals, the expected range for AUC0–∞ or AUC0–t was (0.88–1.12), and for Cmax it was (0.80–1.20).
3 Results
3.1 Demographic Characteristics
Baseline demographics of the enrolled subjects for the SAD and MAD studies are summarized in Table 1. A total of 64 healthy Chinese subjects were enrolled in the SAD study. Fifty-two of those subjects were administered the test drug and 12 received the corresponding placebo treatment. All the subjects completed and were included in the safety and tolerability analysis. The 0.5-mg dose group was only for the assessment of tolerability without collecting pharmacokinetic samples, the 2.5-mg dose group was the pre-test study for pharmacokinetic analysis, and the plasma samples were determined before the method validation. Therefore, subjects from these two dose groups were not included in the pharmacokinetic analysis. Forty-one subjects who received the test drug (one subject discontinued collecting pharmacokinetic samples after receiving the test drug for personal reasons) were included in the pharmacokinetic analysis.
Twenty-four subjects were enrolled in the MAD study, 20 of whom were administered the test drug. All the subjects completed the study as planned and were included in the safety and tolerability analysis. Meanwhile, all study participants who received the test drug (20 subjects) were included in the pharmacokinetic analysis.
3.2 Single-Dose Pharmacokinetics
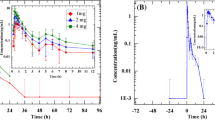
The mean plasma concentration-time profiles of tylerdipine and its metabolites (M2 and M4) following oral administration of 5- to 30-mg doses of tylerdipine hydrochloride are shown in Fig. 2, while the main corresponding pharmacokinetic parameters are summarized in Table 2. After administration, tylerdipine was absorbed with a median Tmax of 1.75–3.00 h, decreasing in a biphasic manner for all doses, with a mean T1/2, 0–72 h of 19.46 h except in the 5-mg dose group (range 18.32–20.66 h). Mean Cmax increased from 0.9993 (5-mg dose group) to 10.11 ng/ml (30-mg dose group); mean AUC0–72 h increased from 4.332 (5-mg dose group) to 73.95 h·ng/ml (30-mg dose group). The apparent volume of distribution (Vz/F) was high (9489 to 40,236 L) suggesting that tylerdipine may be widely distributed in vivo.

Mean (SD) plasma concentration-time profiles of tylerdipine (a) and its metabolites M2 (b) and M4 (c) following oral administration of single ascending doses of tylerdipine hydrochloride in healthy Chinese subjects (5-mg group, n = 8; 10-mg group, n = 8; 15-mg group, n = 8; 20-mg group, n = 8; 25-mg group, n = 5; 30-mg group, n = 4). Inset shows expanded profile of 24 h. LLOQ lower limit of quantification
After oral administration, tylerdipine was metabolized to M2 and M4. As shown in Table 2, the Tmax for M2 and M4 was 2.00–3.50 h (20- to 30-mg groups) and 1.25–3.00 h (5-to 30-mg groups), respectively. Meanwhile, the T1/2 values for M2 and M4 were 7.78–12.57 h (20- to 30-mg groups) and 2.79–5.71 h (5- to 30-mg groups), respectively. The plasma exposure values for M2 and M4 were significantly higher than the parent drug tylerdipine, and the mean fractions of M2 and M4 (calculated as AUC0–t, metabolite/AUC0–t, parent) were 29.3- to 39.2-fold (20- to 30-mg groups) and 13.0- to 20.9-fold (10- to 30-mg groups, 57.1 in the 5-mg group), respectively.
A dose-proportionality analysis of tylerdipine was performed using the power model by WinNonlin software (6.4). The results (Table 3) show that the point estimate and 90% CI for the ratio of AUC0–∞, AUC0–t, and Cmax were 1.70 (1.47–1.94), 1.71 (1.47–1.95), and 1.28 (1.03–1.52), respectively. The results indicated that the increase in AUC0–∞ or AUC0–t was greater than in a dose-proportional manner over the dose range of 5–30 mg, while Cmax exhibited a rough but non-typical dose-proportionality increase.
3.3 Multiple-Dose Pharmacokinetics
Following multiple oral doses of 10 or 20 mg once daily of tylerdipine hydrochloride for 9 consecutive days, the mean plasma concentration-time profiles of tylerdipine and its two metabolites M2 and M4 are depicted in Figs. 3 and 4. The descriptive pharmacokinetic parameters are summarized in Table 4. There were no significant differences in trough plasma concentrations between days 6, 7, and 8 (Fig. 5), indicating that steady-state conditions were achieved after 1 week of daily dosing. Under steady-state conditions, tylerdipine was absorbed with a Tmax ranging from 0.75 to 4.00 on day 9.

Mean (SD) plasma concentration-time profiles of tylerdipine (a) and its metabolites M2 (b) and M4 (c) on Day 1 and Day 9 following oral administration of multiple doses of 10 mg of tylerdipine hydrochloride in healthy Chinese subjects (n = 10). Inset shows expanded profile of 12 h

Mean (SD) plasma concentration-time profiles of tylerdipine (a) and its metabolites M2 (b) and M4 (c) on Day 1 and Day 9 following oral administration of multiple doses of 20 mg of tylerdipine hydrochloride in healthy Chinese subjects (n = 10). Inset shows expanded profile of 12 h

Mean (SD) trough plasma concentrations of tylerdipine (a) and its metabolites M2 (b) and M4 (c) after multiple-dose administration (10 and 20 mg) (n = 10)
For the 10-mg dose, the Cmax values (mean ± SD) for tylerdipine on days 1 and 9 were 2.872 ± 1.180 and 3.031 ± 1.419 ng/ml, respectively. AUC0–24 h values (mean ± SD) were 13.96 ± 3.96 and 23.04 ± 9.94 h·ng/ml, respectively. The accumulation index (mean ± SD) was 1.641 ± 0.511 based on AUC0–24 h and 1.163 ± 0.543 based on Cmax, indicating that there was a slight accumulation of tylerdipine after multiple administrations. Meanwhile, the accumulation indexes (mean ± SD) based on AUC0–24 h and Cmax for M2 were 1.082 ± 0.157 and 0.9622 ± 0.1862, respectively, while corresponding values for M4 were 1.074 ± 0.197 and 0.9505 ± 0.2053, respectively.
For the 20-mg dose, the mean Cmax values (± SD) of tylerdipine on days 1 and 9 were 8.286 ± 5.092 and 11.77 ± 5.65 ng/ml, respectively. Mean AUC0–24 h values (± SD) was 55.90 ± 23.81 and 80.01 ± 34.87 h·ng/ml, respectively. The accumulation index (mean ± SD) was 1.600 ± 0.561 based on AUC0–24 h and 1.629 ± 0.667 based on Cmax. Meanwhile, the accumulation indexes based on AUC0–24 h and Cmax for M2 were 1.125 ± 0.282 and 1.174 ± 0.343, respectively, while the corresponding values for M4 were 1.117 ± 0.325 and 1.221 ± 0.410, respectively.
Based on the AUC accumulation index, the T1/2,eff of tylerdipine was calculated for the 10- and 20-mg dose groups as 18.83 ± 8.52 and 18.37 ± 8.98 h, respectively. After 9 days of once-daily dosing, the mean T1/2 values were 87.23 and 76.26 h, respectively.
3.4 Safety and Tolerability
Tylerdipine hydrochloride appeared to be well tolerated throughout the study, and no serious AEs occurred during the study following administration of single or multiple doses of tylerdipine hydrochloride.
In the SAD study, five AEs were reported in five subjects who received the test drug, including one (in the 20-mg dose group, who experienced toothache) that was considered possibly to be not drug-related and four (one subject in the 5-mg dose group experienced elevated white blood cell count, two subjects in the 15-mg dose group experienced decreased fibrinogen, and one subject in the 20-mg dose group experienced nodal tachycardia) considered to be possibly drug-related by the study investigator. Two subjects in the placebo treatment group reported two AEs, one was toothache considered not to be drug-related and another was elevated white blood cell count in urine considered possibly to be not drug-related. The aforementioned AEs were assessed as mild or moderate in intensity, and subjects recovered without any medical treatment, except for the subjects who experienced toothache, who received concomitant medications for the treatment of the AE. However, none of the subjects were withdrawn from the study due to AEs. Furthermore, there were no clinically significant changes in other subjects, as shown by physical examination, laboratory tests, and ECG reports, before or after the administration of tylerdipine hydrochloride.
In the MAD study, two subjects who received the test drug experienced AEs that were considered to be possibly drug-related by the investigator in the 20-mg dose group. Elevation of creatine kinase (CK), CK-MB, and aspartate transaminase (AST) was observed in one subject after the sixth dose of tylerdipine hydrochloride, which returned to normal with treatment (oral administration of vitamin B1, vitamin C1, and coenzyme Q for 6 days). The other subject experienced elevation of transaminase activities (AST and ALT), recovering without intervention. Except for the two AEs, no clinically significant changes were observed, as shown by physical examination, laboratory tests, and ECG parameters during this study.
4 Discussion
Tylerdipine hydrochloride is a novel dual L/T-type CCB developed for the treatment of hypertension. Here, we report two phase I studies that describe the safety, tolerability, and pharmacokinetics of tylerdipine after oral administration of single and multiple doses in healthy Chinese subjects. These two studies provide the first clinical data for tylerdipine. In both clinical studies, we used two validated LC-MS/MS methods to evaluate the pharmacokinetic properties of tylerdipine and its two metabolites (M2 and M4).
In the SAD study, tylerdipine was absorbed with peak plasma concentrations at approximately 1.75–3.00 h, and the Cmax and AUC increased with rising dose levels from 5 mg to 30 mg. The interindividual variability in Cmax and AUC0–∞ values was considerable, with the coefficients of variation (CVs) ranging from 34.7 to 60.2% and 20.7 to 53.7%, respectively. Tylerdipine was found to be primarily metabolized by CYP3A in in vitro experiments (data not published). Therefore, the high variability observed may be due to the phenotype of isozymes CYP3A4 or CYP3A5. Further investigations of CYP3A4*1G and CYP3A5*3 polymorphisms on the pharmacokinetics of tylerdipine that we analysed indicated that the CYP3A4*1G carriers had a much lower exposure of tylerdipine than those with CYP3A4*1*1 [15]. The results of the dose-proportionality analysis demonstrated that the 90% CIs of AUC0–∞ or AUC0–t for tylerdipine were much higher than the expected 0.88–1.12 interval, indicating that the AUC increased in a greater than dose-proportional manner. Meanwhile, the 90% CI of Cmax was not completely contained within the expected interval, indicating a rough but non-typical dose-proportionality increase. Since the plasma concentrations of tylerdipine were undetectable beyond the 24-h time point in the 5-mg dose group and were up to 1/10 or 1/20 of Cmax at 72 h in other dose groups, the elimination half-life interval of 0–72 h (t1/2, 0–72 h) was further estimated over the dose range of 10–30 mg. Additionally, the mean calculated T1/2, 0–72 h of tylerdipine was 19.46 h, which was shorter than amlodipine (38 h) in plasma [18]. Even so, in vitro studies have demonstrated that tylerdipine exhibits similar or superior antihypertensive effects to amlodipine at the same dose level and also shows organ-protective effects in different animal models (such as SHR, DOCA-salt rats, hemodynamics test in anaesthetized dogs). The pharmacokinetics of tylerdipine may differ in different patient populations. Therefore, future studies are needed to investigate the tolerability, efficacy, and pharmacokinetics in patients with hypertension.
Over the course of the SAD study, in addition to the M4 metabolite, we identified another metabolite, M2, in the 20-mg group. We also determined the concentration of M2 in the following dose groups: After oral administration, tylerdipine was metabolized to M2 and M4. The ratios of the AUC of M2 and M4 to that of tylerdipine were 29.3–39.2 (in the 20- to 30-mg groups) and 13.0–20.9 (in the 10- to 30-mg groups; 57.1 in the 5-mg group), respectively. The results showed that plasma exposure of M2 and M4 was significantly higher than the exposure of tylerdipine. According to the US Food and Drug Administration Guidance for Industry: Safety Testing of Drug Metabolites [19], human metabolites that can raise a safety concern are those formed at greater than 10% of total drug-related exposure at steady state (based on area under the curve). Therefore, the safety of M2 or M4 should be of concern.
In the MAD study, steady-state conditions were achieved after 1 week of daily dosing. In both dose groups, slight accumulation of tylerdipine was observed with a mean Rac of 1.6 based on AUC0–24 h, while the metabolites of M2 and M4 exhibited no accumulation. The concept of an effective half-life (T1/2,eff) of the drug accumulation was applied to the pharmacokinetic data [16]. In principle, T1/2,eff reflects the actual observed drug accumulation rather than one or more aspects of exponential drug disposition, and is calculated based on the AUC accumulation index and the dosing interval [20]. The T1/2,eff (Mean ± SD) for KBP-5660 in the 10- and 20-mg dose groups on Day 9 were 18.83 ± 8.52 and 18.37 ± 8.98 h, respectively. Additionally, mean ± SD values of T1/2 were 87.23 ± 33.72 and 76.26 ± 20.54 h, respectively. Meanwhile, the mean tylerdipine values of T1/2, 0–72 h after multiple doses were longer than those in the single-dose group.
Oral administration of single and multiple doses of tylerdipine hydrochloride was generally well tolerated in healthy Chinese subjects based on the assessment of clinical and laboratory adverse experiences. No serious AEs occurred during the study, and no subject withdrew from study due to AEs.
5 Conclusions
Tylerdipine hydrochloride is generally well tolerated in healthy Chinese subjects. The single ascending-dose study demonstrated that the exposure (AUC) of tylerdipine over the dose range of 5–30 mg increased in a greater than dose-proportional manner, while the Cmax exhibited an approximately dose-proportional increase. Tylerdipine exhibited a slight accumulation following multiple doses in healthy Chinese subjects.
References
Chen BL, Zhang YZ, Luo JQ, Zhang W. Clinical use of azelnidipine in the treatment of hypertension in Chinese patients. Ther Clin Risk Manag. 2015;11:309–17.
Picon RV, Fuchs FD, Moreira LB, Riegel G, Fuchs SC. Trends in prevalence of hypertension in brazil: a systematic review with meta-analysis. PLoS One. 2012;7(10):e48255.
Jarari N, Rao N, Peela JR, Ellafi KA, Shakila S, Said AR, Nelapalli NK, Min Y, Tun KD, Jamallulail SI, Rawal AK, Ramanujam R, Yedla RN, Kandregula DK, Argi A, Peela LT. A review on prescribing patterns of antihypertensive drugs. Clin Hypertens. 2015;22:7.
Wenzel RR. Renal protection in hypertensive patients: selection of antihypertensive therapy. Drugs. 2005;65:29–39.
Zisaki A, Miskovic L, Hatzimanikatis V. Antihypertensive drugs metabolism: an update to pharmacokinetic profiles and computational approaches. Curr Pharm Des. 2015;21:806–22.
Wang AL, Iadecola C, Wang G. New generations of dihydropyridines for treatment of hypertension. J Geriatr Cardiol. 2017;14:67–72.
Dingemanse J, Otasevic P, Shakeri-Nejad K, Klainman E, Putnikovic B, Kracker H, Mueller MS, Zimlichman R. Efficacy and safety of the dual L- and T-type calcium channel blocker, ACT-280778: a proof-of-concept study in patients with mild-to-moderate essential hypertension. J Hum Hypertens. 2015;29:229–35.
Masumiya H, Shijuku T, Tanaka H, Shigenobu K. Inhibition of myocardial L- and T-type Ca2+ currents by efonidipine: possible mechanism for its chronotropic effect. Eur J Pharmacol. 1998;349:351–7.
Ohishi M, Takagi T, Ito N, Terai M, Tatara Y, Hayashi N, Shiota A, Katsuya T, Rakugi H, Ogihara T. Renal-protective effect of T-and L-type calcium channel blockers in hypertensive patients: an Amlodipine-to-Benidipine Changeover (ABC) study. Hypertens Res. 2007;30:797–806.
Ishibashi H, Murai Y, Akaike N. Effect of nilvadipine on the voltage-dependent Ca2+ channels in rat hippocampal CA1 pyramidal neurons. Brain Res. 1998;813:121–7.
Oh IY, Seo MK, Lee HY, Kim SG, Kim KS, Kim WH, Hyon MS, Han KR, Lim SJ, Kim CH. Beneficial effect of efonidipine, an L- and T-type dual calcium channel blocker, on heart rate and blood pressure in patients with mild-to-moderate essential hypertension. Korean Circ J. 2010;40:514–9.
Hayashi K, Wakino S, Sugano N, Ozawa Y, Homma K, Saruta T. Ca2+ channel subtypes and pharmacology in the kidney. Circ Res. 2007;100:342–53.
Hayashi K. L-/T-type Ca channel blockers for kidney protection: ready for sophisticated use of Ca channel blockers. Hypertens Res. 2011;34:910–2.
Hansen PB. Functional importance of T-type voltage-gated calcium channels in the cardiovascular and renal system -news from the world of knock-out mice. Am J Physiol Regul Integr Comp Physiol. 2015;308:227–37.
Zhou S, Tao M, Wang Y, Lu W, Xie L, Chen J, Zhao Y, Yun L, Zhang H, Ning O. Effects of CYP3A4*1G and CYP3A5*3 Polymorphisms on Pharmacokinetics of Tylerdipine hydrochloride in Healthy Chinese Subjects. Xenobiotica. 2018;28:1–6.
Boxenbaum H, Battle M. Effective half-life in clinical pharmacology. J Clin Pharmacol. 1995;35:763–6.
Wang H, Ou N, Lang L, Shi R, Hu P, Jiang J. Pharmacokinetics and pharmacodynamics of intravenous ilaprazole in healthy subjects after single ascending doses. Xenobiotica. 2016;46:1133–41.
Liu MY, Jingying Jia M, Gangyi Liu M, Li S, Chuan LuB, Yanmei Liu M, Yu MC. Pharmacokinetics and bioequivalence evaluation of two formulations of 10-mg amlodipine besylate: An open-label, single-dose, randomized, two-way crossover study in healthy chinese male volunteers. Clin Ther. 2009;31:777–83.
US Food and Drug Administration. Safety testing of drug metabolites guidance for industry. 2016. http://www.fda.gov/downloads/drugs/guidancecompliance regulatoryinformation/guidances/ucm079266.pdf. Accessed 22 Nov 2016.
Jiang J, Li L, Yin H, Woessner R, Emotte C, Li R, Khindri S, Pei H. Single- and multiple-dose pharmacokinetics of inhaled indacaterol in healthy Chinese volunteers. Eur J Drug Metab Pharmacokinet. 2015;40:203–8.
Acknowledgement
The authors thank all the subjects who participated in the studies. This work was supported by the XuanZhu Pharma Co., Ltd. (Jinan, China).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Xuan Zhu Pharma Co., Ltd. (Jinan, China).
Conflict of interest
The authors have no potential conflicts of interest to report.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The studies were approved by the Ethics Committee of the First Affiliated Hospital with Nanjing Medical University (Nanjing, China) and the approval numbers are 2014-MD-164.A1 for SAD study and 2015-MD-141 for MAD study, respectively.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Zhou, S., Wang, Y., Wang, L. et al. Pharmacokinetics, Safety and Tolerability of Tylerdipine Hydrochloride, a Novel Dihydropyridine Dual L/T-type Calcium Channel Blocker, after Single and Multiple Oral Doses in Healthy Chinese Subjects. Clin Drug Investig 39, 85–96 (2019). https://doi.org/10.1007/s40261-018-0722-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-018-0722-5