
Abstract
Background
Gastroprotective agents are recommended for patients receiving low-dose aspirin (LDA) or nonsteroidal anti-inflammatory drugs (NSAIDs). Vonoprazan is a potassium-competitive acid blocker recently approved for the prevention of peptic ulcer recurrence in patients receiving LDA or NSAIDs.
Methods
This phase 2, open-label, single-center study in healthy Japanese males evaluated drug–drug interactions between vonoprazan 40 mg and LDA (100 mg) or NSAIDs [loxoprofen sodium (60 mg), diclofenac sodium (25 mg), or meloxicam (10 mg)] and vice versa. Subjects were allocated to one of eight cohorts and received their orally administered treatment regimen (to assess the effect of vonoprazan vs. NSAID or LDA, or vice versa) once daily. Endpoints were the pharmacokinetics of plasma concentrations of the study drugs alone and in combination (primary), safety (secondary), and vonoprazan effects on aspirin-mediated inhibition of platelet-aggregation.
Results
Of 109 subjects screened, 64 were assigned to one of eight cohorts (n = 8 per cohort) and received treatment, one subject discontinued due to a treatment-emergent adverse event (TEAE), and 63 completed the study. There were few differences in the pharmacokinetics of vonoprazan when administered with LDA or NSAIDs, and few differences in the pharmacokinetics of LDA or NSAIDs when administered with vonoprazan. The differences were small and not clinically meaningful. Inhibition of arachidonic-induced platelet aggregation by LDA was not influenced by vonoprazan. Six patients experienced a TEAE, all were mild and were deemed unrelated to study drugs. One subject withdrew due to infection (tonsillitis).
Conclusions
No clinically meaningful drug–drug interactions were observed and vonoprazan was well tolerated when administered with LDA or NSAIDs.
Study registration
JapicCTI-153100
Similar content being viewed by others


Avoid common mistakes on your manuscript.
There were no clinically meaningful differences in the pharmacokinetics of vonoprazan when administered concomitantly with aspirin, loxoprofen sodium, diclofenac sodium, or meloxicam. |
There were no clinically meaningful differences in the pharmacokinetics of aspirin, loxoprofen sodium, diclofenac sodium, or meloxicam when administered concomitantly with vonoprazan. |
This study shows that vonoprazan is not associated with drug–drug interactions between low-dose aspirin or the nonsteroidal anti-inflammatory drugs loxoprofen sodium, diclofenac sodium, or meloxicam. |
1 Introduction
Patients receiving long-term administration of low-dose aspirin (LDA) or nonsteroidal anti-inflammatory drugs (NSAIDs) are at increased risk of developing gastric or duodenal ulcers [1]. Therefore guidelines throughout the world recommend that patients receiving LDA or NSAIDs also receive proton pump inhibitors (PPIs), histamine H2-receptor antagonists (H2RAs), or prostaglandin analogs to reduce gastric acid levels [2–6]. The efficacy of these agents is variable, but the agents of choice are PPIs [3–5]. In 2010, lansoprazole was the first PPI approved for this indication in Japan. Ulcer recurrence in some patients (12.7 % of patients with NSAID-induced ulcers [7] and 3.7 % with LDA-induced ulcers [8]) occurs despite lansoprazole treatment indicating that secondary prevention is incomplete. Hence, a more effective agent is needed [7, 8].
Vonoprazan, an orally active potassium-competitive acid blocker (P-CAB), effectively reduces gastric acid and has recently been approved in Japan for the treatment of several acid-related disease conditions [9–11]. Vonoprazan is a P-CAB that ionically and reversibly binds to the gastric proton pump [12, 13]. Unlike previously-developed P-CABs, vonoprazan is a different chemotype and therefore avoids the hepatotoxicity seen with previous P-CABs [13–16]. Moreover, unlike PPIs, vonoprazan does not require intermediate complex formation, is stable at low pH, and its bioavailability is unaffected by food [11, 13]. Vonoprazan is rapidly absorbed in healthy adult males, the time to reach the maximum observed plasma concentration (T max) is ≤2 h and the mean elimination half-life is 9 h [17]. In vitro studies indicate that vonoprazan is extensively metabolized by multiple metabolizing enzymes in humans including cytochrome P450 (CYP) 3A4/5, CYP2C19, CYP2C9, and the non-CYP enzyme sulfotransferase (SULT)2A1 [18]. CYP3A4 predominantly contributes to the metabolism of vonoprazan to M-I, M-III, and N-demethylated vonoprazan, while SULT2A1 contributes to the metabolism of vonoprazan to form vonoprazan N-sulfate, and CYP2C9 mediates the formation of M-IV-Sul from vonoprazan N-sulfate. Metabolites M-I, M-II, M-III, and M-IV-Sul were quantified in human plasma and none of them are pharmacologically active.
Vonoprazan is effective in promoting healing of erosive esophagitis and gastric/duodenal ulcers, and as an adjunct to Helicobacter pylori eradication [9, 10, 11]. Additionally, vonoprazan is useful in preventing recurrence of both erosive esophagitis and of LDA- or NSAID-induced gastric/duodenal ulcers [9, 11].
The objectives of this study were to evaluate the effect of multiple doses of LDA and the most commonly used NSAIDs in Japan (loxoprofen, diclofenac, and meloxicam) on the pharmacokinetics of vonoprazan and, conversely, to evaluate the effects of multiple doses of vonoprazan on the pharmacokinetics of LDA or common NSAIDs. Additionally, the study was designed to evaluate the safety of vonoprazan administered in combination with LDA or NSAIDs, and the effects of vonoprazan on aspirin-mediated inhibition of platelet aggregation.
2 Subjects and Methods
2.1 Study Design
This was a single-site, phase 2, open-label, one-way crossover, clinical pharmacology study (JapicCTI-153100). The pharmacokinetics of vonoprazan were comparatively evaluated in healthy Japanese male subjects when vonoprazan (40 mg) was administered alone or in combination with LDA (100 mg) or NSAIDs [loxoprofen sodium (180 mg), diclofenac sodium (75 mg), or meloxicam (10 mg)]. The pharmacokinetics of aspirin or NSAIDs were evaluated when aspirin or NSAIDs were administered alone and in combination with vonoprazan (Fig. 1).

Study design for Cohorts 1–4, 6, and 7 (a), Cohort 5 (b), and Cohort 8 (c)
2.2 Study Population
Healthy Japanese male subjects were screened using predefined inclusion and exclusion criteria. Inclusion criteria were: age 20–45 years; body weight ≥50 kg; body mass index between 18.5 and 24.9 kg/m2; negative for hepatitis B, hepatitis C, human immunodeficiency virus, and syphilis. For subjects in Cohort 5 (measuring pharmacokinetic effect of vonoprazan on aspirin and its metabolite), an additional inclusion criterion was platelet-aggregating activity of ≥70 % induced by collagen or arachidonic acid.
Exclusion criteria were: medical history of hepatic, renal, cardiovascular, hematological, endocrine, metabolic, pulmonary, gastrointestinal, neurological, urological, immunological, or psychiatric disease that would preclude eligibility to participate; allergy or hypersensitivity to any drugs or food; history of drug or alcohol abuse within the past 5 years; poor venous access; history of blood collection of at least 200 ml (within 4 weeks); 400 ml (within 12 weeks), or 800 ml (within 52 weeks); history of blood component collection (within past 2 weeks); use of prescription drugs, vitamins, Chinese herbal remedies, or supplements within the past 4 weeks; ingested food or beverages containing grapefruit, caffeine, or alcohol within the past 72 h; participation in a drug trial in the past 16 weeks; or participation in a previous clinical study of vonoprazan. Subjects were free to withdraw from the study at any time without having to provide a reason.
2.3 Treatment Protocol
The study consisted of eight cohorts comprising eight subjects each (Table 1). The treatment phase consisted of a single dose of the substrate (Medication A), initially administered alone, followed by a suitable washout period (2 days for Cohort 1–4, 6, and 7, 13 days for Cohort 5, and 4 days for Cohort 8) and then a 6-day period during which multiple doses of the interacting drug (Medication B) were administered. Medication A was concomitantly administered on the fifth day of the 6-day multiple-dose period (Fig. 1a–c).
Washout periods were determined based on the elimination half-life of each study medication, where the minimum wash-out period chosen in each cohort was a minimum of five times the half-life of each medication.
For plasma analysis, blood was collected 0.5 h before and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, and 12 h after study drug administration [Day 1, Day 8 (excluding Cohort 5), Day 19 (Cohort 5)], at 9:00 am (Days 2–7), and 16, 24, and 48 h after drug administration (Days 9–10); one blood sample was collected on Day 15 (excluding Cohort 5). For Cohort 5, blood was collected at 9:00 am (Days 15–18) and 16, 24, and 48 h after drug administration (Days 20–21); one blood sample for Cohort 5 was collected on Day 26. Plasma was separated at 4 °C by centrifugation (1500g for 10 min) and then stored at −80 °C. Plasma concentrations of analytes were determined using a validated method of liquid chromatography tandem mass spectrometry. The lower limits of quantification (LLOQ) of vonoprazan, M-I, M-II, M-III, and M-IV-Sul were 0.1, 1, 1, 0.1, and 0.1 ng/ml, respectively. The LLOQ of aspirin, salicylic acid, loxoprofen, trans-OH loxoprofen, diclofenac, and meloxicam were 2, 100, 10, 2, 1, and 3 ng/ml, respectively.
2.4 Outcome Measures
2.4.1 Assessment of Pharmacokinetic Variables
Plasma concentrations of the following analytes were determined in order to identify potential drug–drug interactions: vonoprazan free-base (vonoprazan-F) and its metabolites (M-I, M-II, M-III, and M-IV-Sul), unchanged aspirin and its metabolite (salicylic acid), unchanged loxoprofen and its active metabolite (trans-OH metabolite), unchanged diclofenac, and unchanged meloxicam.
Pharmacokinetic measures included the area under the concentration-time curve from time 0 to time of last quantifiable concentration (AUC0–last), calculated using the linear trapezoidal rule and the terminal elimination rate constant (λz), calculated as the negative of the slope of the log-linear regression of the natural logarithm concentration-time curve during the terminal phase. Other pharmacokinetic parameters analyzed included the maximum observed concentration (C max); the time to reach C max (T max); the area under the concentration-time curve from time 0 to infinity, calculated as AUC0–inf = AUC0–last + C last/λz, where last is the time of the last quantifiable concentration, and C last is the last quantifiable concentration; the terminal elimination half-life (T 1/2), calculated as T 1/2 = ln(2)/λz, and the apparent clearance after extravascular administration (CL/F), calculated as CL/F = Dose/AUC0–inf after a single dose and as CL/F = Dose/AUC0–tau after multiple dosing (at steady state).
2.4.2 Assessment of Pharmacodynamic Effects
The effect of vonoprazan on the platelet-aggregating inhibitory activity of aspirin was evaluated in Cohort 5. A 4.5-ml aliquot of blood was collected to determine platelet-aggregating activity [maximum aggregation (% transmission)] using 1 µg/ml of collagen and 1 mmol/l of arachidonic acid as platelet-aggregation-inducing agents. Platelet aggregation was measured using the P200 aggregometer (Kowa Inc, Nagoya, Japan) according to Ozaki et al. [19]. On Day 1 and Day 19, blood samples were collected 0.5 h before and 1.5, 4, 6, 8, 10, and 12 h after study drug administration. On Days 2, 3, 20, and 21, blood samples were collected at 9:00 am.
2.4.3 Assessment of Safety Variables
Safety variables included treatment-emergent adverse events (TEAEs), serious adverse events (SAEs), vital signs, weight, electrocardiogram (ECG) findings, and clinical laboratory tests (hematology, blood chemistry, and urinalysis). An adverse event (AE) was defined as any untoward medical occurrence after administration of a pharmaceutical product; AEs were graded as mild, moderate, or severe. AEs did not necessarily have to have a causal relationship with treatment. A TEAE was defined as an AE with the date of onset that occurred after receiving study drug through study discharge, or a continuing AE diagnosed before the date of first administration of study drug, which increased in intensity or frequency after the start of dosing. A SAE was defined as any occurrence that was life threatening, or resulted in death, hospitalization, significant disability or incapacity, or a congenital anomaly or birth defect.
2.4.4 Assessment of Helicobacter pylori Status
Participants were screened for H. pylori antibody using a serum antibody test. Values <10 U/ml were considered negative and those ≥10 U/ml were considered positive.
2.5 Ethics
The institutional review board for SOUSEIKAI Hakata Clinic was responsible for the initial and continuing review and approval of the clinical study in accordance with the requirements of Good Clinical Practice. The protocol and the subject informed consent form for this study were approved in writing before commencement of the study.
2.6 Statistical Analysis
No sample size calculation was performed. The sample sizes were determined after considering the feasibility of the study. A statistical analysis plan was prepared and finalized before database lock. The pharmacokinetic analysis set comprised subjects who received the study drug, had no significant protocol deviations, completed the minimum required study procedures, and had sufficient drug concentration data. The safety analysis set was defined as all subjects who received at least one dose of study drug.
Concentrations below the quantification limit were treated as 0 in the computation of summary statistics. Missing values due to discontinuation or lack of measurement were not interpolated and were excluded from the summary statistics.
Pharmacokinetic parameters were derived for single- and combination-dose periods from concentrations of each analyte of Medication A up to 48 h after dosing by noncompartmental analysis using WinNonlin V5.3 (Pharsight—A Certara Company, Princeton, NJ, USA).
Summary statistics (number of subjects, mean, standard deviation, standard error, maximum, minimum and quartile value) were used to summarize concentrations of each analyte at each time point and the pharmacokinetic parameters. Analysis of variance was performed on the natural logarithms of AUC0–inf, AUC0–48, and C max of Medication A as dependent variables; dosing conditions (combination dose period or single dose period) were fixed effects, in order to examine the drug–drug interaction. A point estimate and 90 and 95 % confidence intervals (CIs) of the least squares (LS) means ratios between dosing conditions (combination dose period–single dose period) were provided. The 90 % CIs were compared with the standard bioequivalence criteria of 0.8 to 1.25 [20]. Non-transformed parameters were also analyzed for reference.
Summary tables were generated for TEAEs by preferred term for each treatment period (single dose period, multiple dose period, combination dose period).
3 Results
3.1 Demographic and Baseline Clinical Characteristics
A total of 109 subjects were screened. Of these, 64 subjects were entered into the study (Fig. 2). The reasons for screening failure were voluntary withdrawal or did not meet entrance criteria. Sixty-three subjects completed the study. No clinically relevant differences were observed among the cohorts for any demographic characteristics at baseline (Table 2). Subjects had a mean age of 24.2 years, most (79.7 %) were negative for H. pylori antibody.

Patient disposition. aIncluding four subjects who were judged “eligible” at screening. bOne subject was replaced before administration because a more appropriate subject was identified
3.2 Pharmacokinetic Measures
Of the 64 patients enrolled, 61 comprised the pharmacokinetic analysis set. Three patients were excluded (one each in Cohorts 1, 4, and 7) due to lack of samples for pharmacokinetic evaluation.
3.2.1 Vonoprazan-F and its Metabolites (Cohorts 1–4)
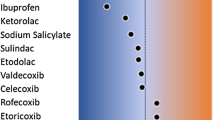
Few or no differences were observed in the pharmacokinetics of vonoprazan-F or its metabolites when vonoprazan was administered alone or concomitantly with aspirin (Fig. 3, Supplementary Table 1). The 90 % CIs of the ratio of LS means for C max and AUCs of vonoprazan-F were within the range (0.8, 1.25), except that the upper bound for C max was 1.257, marginally above 1.25. Slight increases (approximately 13–24 %) were observed in C max and AUCs of M-II when vonoprazan was administered concomitantly with aspirin. The 90 % CIs of the ratios of LS means for C max and AUCs of M-II were not within the range (0.8, 1.25).

Effect of aspirin, loxoprofen sodium, diclofenac sodium, or meloxicam on the pharmacokinetics of vonoprazan-F when coadministered. Ratio of least square means and 90 % confidence intervals (CIs) are presented. Dotted lines represent the standard equivalence criterion of 0.80 to 1.25 rate limits for the 90 % CIs. AUC area under the plasma concentration-time curve, AUC 0–48 AUC from time 0–48 h, calculated using the linear trapezoid rule, AUC 0–inf AUC from time 0 to infinity, C max maximum observed plasma concentration
No differences were observed in C max and AUCs of M-I, M-III, and M-IV-Sul when vonoprazan was administered alone or concomitantly with aspirin. The 90 % CIs of the ratios of LS means for C max and AUCs for the three metabolites were within the range (0.8, 1.25).
Few or no differences were observed in the pharmacokinetics of vonoprazan-F or its metabolites when vonoprazan was administered alone or concomitantly with loxoprofen (Fig. 3, Supplementary Table 2). The 90 % CIs of the ratios of LS means for C max and AUCs of vonoprazan-F were within the range (0.8, 1.25), except that the lower bound for C max was 0.695, marginally below 0.80. Few or no differences in M-I, M-II, and M-IV-Sul were observed in C max and AUCs when vonoprazan was administered alone or concomitantly with loxoprofen sodium. The 90 % CIs of the ratios of LS means for C max and AUCs were within the range (0.8, 1.25), except that the lower bounds for the C max of M-I and M-IV-Sul were 0.787 and 0.772, respectively, marginally below 0.80, and the upper bound for AUC0–inf of M-II was 1.258, marginally above 1.25. No differences were observed in C max and AUCs of M-III when vonoprazan was administered alone or concomitantly with loxoprofen sodium. The 90 % CIs of the ratios of LS means for C max and AUCs of M-III were within the range (0.8, 1.25).
Few or no differences were observed in the pharmacokinetics of vonoprazan-F when vonoprazan was administered alone or concomitantly with diclofenac sodium (Fig. 3, Supplementary Table 3). The 90 % CIs of the ratios of LS means for C max and AUCs of vonoprazan-F were within the range (0.8, 1.25). A slight decrease (approximately 13 %) was observed in the C max of M-I when vonoprazan was administered concomitantly with diclofenac sodium. The 90 % CIs of the ratios of LS means for C max and AUCs for M-I were within the range (0.8, 1.25), except that the lower bound for C max was 0.759, below 0.80. A slight decrease (approximately 12 %) was observed in AUC0–inf of M-II when vonoprazan was administered concomitantly with diclofenac sodium. The 90 % CIs of the ratios of LS means for C max and AUCs for M-II were within the range (0.8, 1.25), except that the lower bound for AUC0–inf was 0.682, below 0.80. No differences in M-III and M-IV-Sul were observed in C max and AUCs when vonoprazan was administered alone or concomitantly with diclofenac sodium. The 90 % CIs of the ratios of LS means for C max and AUCs were within the range (0.8, 1.25).
No differences were observed in the pharmacokinetics of vonoprazan-F when vonoprazan was administered alone or concomitantly with meloxicam (Fig. 3, Supplementary Table 4). The 90 % CIs of the ratios of LS means for C max and AUCs of vonoprazan-F were within the range (0.8, 1.25). A slight increase (approximately 12 %) was observed in AUC0–inf of M-II, when vonoprazan was administered concomitantly with meloxicam. The 90 % CIs of the ratios of LS means for C max and AUCs were within the range (0.8, 1.25), except that the upper bound for AUC0–inf was 1.255, slightly above 1.25. No differences in M-I, M-III, and M-IV-Sul were observed in C max and AUCs when vonoprazan was administered alone or concomitantly with meloxicam. The 90 % CIs of the ratios of LS means for C max and AUCs for these three metabolites were within the range (0.8, 1.25).
3.2.2 Aspirin and its Metabolites (Cohort 5)
An 83 % increase was observed in C max and a slight increase (approximately 18 %) was observed in AUC0–48 of aspirin, when aspirin was administered concomitantly with vonoprazan (Fig. 4, Supplementary Table 5). The 90 % CIs of the ratios of the LS means for C max and AUCs were not within the range (0.80, 1.25). One subject exhibited very slow absorption (T max was 24 h); additional analyses excluding this patient were performed. After exclusion of this subject, an approximately 56 % increase was observed in C max and a slight increase (approximately 23 %) was observed in the AUC0–48 of aspirin when aspirin was administered concomitantly with vonoprazan (Fig. 4). The 90 % CIs of the ratios of LS means for C max and AUCs were not within the range (0.80, 1.25).

Effect of vonoprazan on the pharmacokinetics of aspirin (full cohort and cohort excluding one patient who exhibited very slow absorption), loxoprofen sodium, diclofenac sodium, or meloxicam when coadministered. Ratio of least square means and 90 % confidence intervals (CIs) are presented. Dotted lines represent the standard equivalence criterion of 0.80 to 1.25 rate limits for the 90 % CIs. AUC0–inf for aspirin + vonoprazan is not shown because it was not possible to estimate the elimination rate constant for five subjects in the single-dose period and three subjects in the combination dose period due to insufficient data in the elimination phase. AUC area under the plasma concentration-time curve, AUC 0–48 AUC from time 0–48 h, calculated using the linear trapezoid rule, AUC 0–inf AUC from time 0 to infinity, C max maximum observed plasma concentration
Few differences were observed in the pharmacokinetics of salicylic acid, when aspirin was administered alone or concomitantly with vonoprazan (Supplementary Table 5). Though the 90 % CIs of the ratios of LS means for C max and AUCs were not within the range (0.80, 1.25), the point estimate for C max was around 1, and the lower bounds of AUCs were 0.765 to 0.783, marginally below 0.80. This small difference was not clinically meaningful.
3.2.3 Loxoprofen and its Metabolites (Cohort 6)
No differences were observed in the pharmacokinetics of loxoprofen or its trans-OH metabolite when loxoprofen sodium was administered alone or concomitantly with vonoprazan (Fig. 4, Supplementary Table 6). The 90 % CIs of the ratios of LS means for C max and AUCs were within the range (0.80, 1.25) for both loxoprofen and its trans-OH metabolite.
3.2.4 Diclofenac Sodium (Cohort 7)
A slight increase (approximately 27 %) was observed in the C max of diclofenac when diclofenac sodium was administered alone or concomitantly with vonoprazan (Fig. 4, Supplementary Table 7). However, few or no differences were observed in other pharmacokinetic parameters. The 90 % CIs of the ratios of LS means for C max and AUCs were within the range (0.80, 1.25), except that the upper bound for C max was 1.764, above 1.25. This significant difference was not clinically meaningful, because the point estimate for C max was 1.274 and the lower bound for C max was 0.920.
3.2.5 Meloxicam (Cohort 8)
Although a slight increase (approximately 18 %) was observed in the AUC0–inf of meloxicam, few or no differences were observed in other pharmacokinetic parameters when meloxicam was administered alone or administered concomitantly with vonoprazan (Fig. 4, Supplementary Table 8). The 90 % CIs of the ratios of LS means for C max and AUCs were within the range (0.80, 1.25), except that the upper bound for AUC0–inf was 1.263, marginally above 1.25. This small difference was not clinically meaningful.
3.3 Pharmacodynamic Measures
No differences were observed in arachidonic acid-induced platelet-aggregating activity when aspirin was administered alone or concomitantly with vonoprazan (Fig. 5b, d). Concomitant vonoprazan slightly reduced the collagen-induced platelet-aggregating inhibitory activity of aspirin (Fig. 5a, c).

Pharmacodynamic effects of vonoprazan on a collagen-induced or b arachidonic acid-induced platelet-aggregation activity during coadministration in the full cohort. Pharmacodynamic effects of vonoprazan on c collagen-induced or d arachidonic acid-induced platelet-aggregation activity during coadministration in the cohort excluding one patient who exhibited very slow absorption. Means and standard deviations are shown
3.4 Safety and Tolerability Measures
Among 64 subjects, a total of six subjects experienced TEAEs during this study (Table 3). All TEAEs were mild in intensity and not related to study drugs. The TEAEs were observed in Cohorts 2, 5, 6, and 7. In Cohort 2, pharyngitis occurred in one subject when vonoprazan was administered alone. In Cohort 5, tonsillitis that led to discontinuation occurred in one subject when aspirin was administered concomitantly with vonoprazan. In Cohort 6, blood creatine phosphokinase increased occurred in two subjects when loxoprofen sodium was administered concomitantly with vonoprazan. In Cohort 7, epistaxis occurred in one subject when diclofenac sodium was administered alone and alanine aminotransferase increase occurred in one subject when diclofenac sodium was administered concomitantly with vonoprazan.
No serious TEAEs were reported and no deaths occurred during the study period. No instances of serious liver dysfunction were reported during the study. All of the TEAEs occurred during the combination dose period (i.e., loxoprofen sodium with vonoprazan in Cohort 6 and diclofenac sodium with vonoprazan in Cohort 7). No hematology or urinalysis result or ECG finding was assessed by the investigator or subinvestigator to be clinically significant or a TEAE.
4 Discussion
This study is the first to examine the potential risk for drug–drug interactions between vonoprazan and LDA or vonoprazan and NSAIDs. The results of this study showed that there were no clinically significant effects of LDA or the NSAIDs loxoprofen sodium, diclofenac sodium, and meloxicam on the pharmacokinetics of vonoprazan, and no clinically significant effects of vonoprazan on the pharmacokinetics of LDA or the NSAIDs.
Long-term LDA or NSAIDs have been associated with increased risk of upper gastrointestinal events such as ulcers and bleeding [21]. Therefore, acid-suppressive agents are concomitantly administered to provide gastroprotection [21]. Hence, it was important to assess whether vonoprazan was associated with drug–drug interactions with LDA or commonly prescribed NSAIDs. Loxoprofen sodium, diclofenac sodium, and meloxicam are the most commonly prescribed NSAIDs in Japan for the management of pain in patients with osteoarthritis or rheumatoid arthritis [22]. The results of this study support the lack of drug–drug interactions between vonoprazan and LDA or these NSAIDs.
Low-dose aspirin is a key component of thromboprophylaxis in patients at risk of adverse cardiovascular events such as myocardial infarction, angina pectoris, or atrial fibrillation (approved indication outside Japan). Therefore, it was important to confirm the lack of pharmacokinetic and pharmacodynamic effects of vonoprazan on aspirin. The effects of vonoprazan on LDA and salicylic acid pharmacokinetics were difficult to determine because of the large inter-subject variability observed. Although the AUCs of aspirin increased when concomitantly administered with vonoprazan, few differences were observed in the pharmacokinetics of salicylic acid. Similarly, variability in the pharmacodynamic effects of aspirin were also observed in that vonoprazan slightly reduced the collagen-induced platelet-aggregating inhibitory activity of aspirin during concomitant administration. However, the arachidonic acid-induced platelet-aggregating inhibitory activity of aspirin was not influenced by vonoprazan administration. In addition, this study used a low dose of aspirin which is within the range commonly used to prevent thromboembolism (100 mg), whereas the maximum dose is 324 mg. Previous studies have reported that an increased concentration of salicylic acid has no meaningful effect on platelet aggregation [23, 24]. Overall, vonoprazan was considered to have no significant effect on the pharmacokinetics or pharmacodynamics of aspirin and salicylic acid and therefore can be considered to have no clinically relevant drug–drug interactions when taken in combination with LDA. In addition, concomitant administration of vonoprazan with LDA or NSAIDs was well tolerated. Few AEs were noted during the study.
The strengths of this study include the prospective design and the high study completion rate. However, because this was a clinical pharmacology study, the sample size was small and there was a limited duration of follow-up. The subjects enrolled in the study were relatively young whereas patients with osteoarthritis or rheumatoid arthritis are likely to be older. In addition, the study was conducted only in healthy male volunteers in order to compare the results with other pharmacokinetic studies of vonoprazan. Although caution is advised when extrapolating to women, gender-related disparities in pharmacokinetics are largely inconsequential [25]. Therefore, the potential for drug–drug interactions cannot be ruled out in females or in an older population who may be taking other concomitant medications.
5 Conclusion
In conclusion, despite some small differences, no clinically meaningful drug–drug interactions were observed in healthy male volunteers between vonoprazan and either LDA or commonly prescribed NSAIDs. The results suggest that vonoprazan at the dose evaluated in this study can be concomitantly administered with LDA or NSAIDs such as loxoprofen sodium, diclofenac sodium, or meloxicam.
References
McCarberg BH, Cryer B. Evolving therapeutic strategies to improve nonsteroidal anti-inflammatory drug safety. Am J Ther. 2015;22(6):e167–78.
Chou R, Helfand M, Peterson K, Dana T, Roberts C. Comparative effectiveness and safety of analgesics for osteoarthritis. Comparative effectiveness review No. 4. Rockville: Agency for Healthcare Research and Quality; 2006.
Japanese Society of Gastroenterology. Guideline for peptic ulcer treatment [in Japanese]. Tokyo: Nankodo K.K; 2009.
Lanza FL, Chan FK, Quigley EM. Practice Parameters Committee of the American College of G. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104(3):728–38.
Research Group for Assessment and Adaptation of Guideline for Gastric Ulcer. Evidence-based guideline for gastric ulcer treatment [in Japanese]. 2nd ed. Tokyo: Jiho K.K; 2007.
Rostom A, Dube C, Wells G, Tugwell P, Welch V, Jolicoeur E, et al. Prevention of NSAID-induced gastroduodenal ulcers. Cochrane Database Syst Rev. 2002;4:CD002296.
Sugano K, Kontani T, Katsuo S, Takei Y, Sakaki N, Ashida K, et al. Lansoprazole for secondary prevention of gastric or duodenal ulcers associated with long-term non-steroidal anti-inflammatory drug (NSAID) therapy: results of a prospective, multicenter, double-blind, randomized, double-dummy, active-controlled trial. J Gastroenterol. 2012;47(5):540–52.
Sugano K, Matsumoto Y, Itabashi T, Abe S, Sakaki N, Ashida K, et al. Lansoprazole for secondary prevention of gastric or duodenal ulcers associated with long-term low-dose aspirin therapy: results of a prospective, multicenter, double-blind, randomized, double-dummy, active-controlled trial. J Gastroenterol. 2011;46(6):724–35.
Ashida K, Sakurai Y, Hori T, Kudou K, Nishimura A, Hiramatsu N, et al. Randomised clinical trial: vonoprazan, a novel potassium-competitive acid blocker, vs. lansoprazole for the healing of erosive oesophagitis. Aliment Pharmacol Ther. 2016;43(2):240–51.
Murakami K, Sakurai Y, Shiino M, Funao N, Nishimura A, Asaka M. Vonoprazan, a novel potassium-competitive acid blocker, as a component of first-line and second-line triple-therapy for Helicobacter pylori eradication. Gut. 2016. doi:10.1136/gutjnl-2015-311304 (ePub ahead of print).
Takeda Takecab (vonoprazan tablets) package insert. Available at: http://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/400256_2329030F1020_1_04. Accessed 9 Aug 2016.
Hori Y, Imanishi A, Matsukawa J, Tsukimi Y, Nishida H, Arikawa Y, et al. 1-[5-(2-Fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamin e monofumarate (TAK-438), a novel and potent potassium-competitive acid blocker for the treatment of acid-related diseases. J Pharmacol Exp Ther. 2010;335(1):231–8.
Shin JM, Inatomi N, Munson K, Strugatsky D, Tokhtaeva E, Vagin O, et al. Characterization of a novel potassium-competitive acid blocker of the gastric H, K-ATPase, 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamin e monofumarate (TAK-438). J Pharmacol Exp Ther. 2011;339(2):412–20.
Berg AL, Bottcher G, Andersson K, Carlsson E, Lindstrom AK, Huby R, et al. Early stellate cell activation and veno-occlusive-disease (VOD)-like hepatotoxicity in dogs treated with AR-H047108, an imidazopyridine proton pump inhibitor. Toxicol Pathol. 2008;36(5):727–37.
Dent J, Kahrilas PJ, Hatlebakk J, Vakil N, Denison H, Franzen S, et al. A randomized, comparative trial of a potassium-competitive acid blocker (AZD0865) and esomeprazole for the treatment of patients with nonerosive reflux disease. Am J Gastroenterol. 2008;103(1):20–6.
Kahrilas PJ, Dent J, Lauritsen K, Malfertheiner P, Denison H, Franzen S, et al. A randomized, comparative study of three doses of AZD0865 and esomeprazole for healing of reflux esophagitis. Clin Gastroenterol Hepatol. 2007;5(12):1385–91.
Jenkins H, Sakurai Y, Nishimura A, Okamoto H, Hibberd M, Jenkins R, et al. Randomised clinical trial: safety, tolerability, pharmacokinetics and pharmacodynamics of repeated doses of TAK-438 (vonoprazan), a novel potassium-competitive acid blocker, in healthy male subjects. Aliment Pharmacol Ther. 2015;41(7):636–48.
Yamasaki H, Kawaguchi N, Nonaka M, Takahashi J, Morohashi A, Hirabayashi H et al. In vitro metabolism of TAK-438, vonoprazan fumarate, a novel potassium-competitive acid blocker. Xenobiotica. 2016. doi:10.1080/00498254.2016.1203505 (ePub ahead of print).
Ozaki Y, Satoh K, Yatomi Y, Yamamoto T, Shirasawa Y, Kume S. Detection of platelet aggregates with a particle counting method using light scattering. Anal Biochem. 1994;218(2):284–94.
English translation of Attachment 1 of Pharmaceutical and Food Safety Bureau/Evaluation and Licensing Division Notification No. 1124004. [Guideline for Bioequivalence Studies of Generic Products]. 2016. http://www.nihs.go.jp/drug/be-guide(e)/be2006e.pdf. Accessed 25 Aug 2016.
Goldstein JL, Cryer B. Gastrointestinal injury associated with NSAID use: a case study and review of risk factors and preventative strategies. Drug Healthc Patient Saf. 2015;7:31–41.
Sugano K, Kinoshita Y, Miwa H, Takeuchi T, Esomeprazole NPSG. Safety and efficacy of long-term esomeprazole 20 mg in Japanese patients with a history of peptic ulcer receiving daily non-steroidal anti-inflammatory drugs. BMC Gastroenterol. 2013;13:54.
Roberts MS, McLeod LJ, Cossum PA, Vial JH. Inhibition of platelet function by a controlled release acetylsalicylic acid formulation–single and chronic dosing studies. Eur J Clin Pharmacol. 1984;27(1):67–74.
Ross-Lee LM, Elms MJ, Cham BE, Bochner F, Bunce IH, Eadie MJ. Plasma levels of aspirin following effervescent and enteric coated tablets, and their effect on platelet function. Eur J Clin Pharmacol. 1982;23(6):545–51.
Meibohm B, Beierle I, Derendorf H. How important are gender differences in pharmacokinetics? Clin Pharmacokinet. 2002;41(5):329–42.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding Support
This study was sponsored by Takeda Pharmaceutical Company Limited, manufacturer/licensee of vonoprazan. Medical writing assistance was provided by Elise Magatova, PhD, Tania Dickson, PhD, and Serina Stretton, PhD, CMPP of ProScribe – Envision Pharma Group, and was funded by Takeda Pharmaceutical Company Limited. ProScribe’s services complied with international guidelines for Good Publication Practice (GPP3).
Role of the Sponsor
Takeda Pharmaceutical Company Limited was involved in the study design, data collection, data analysis, and preparation of the manuscript.
Role of Contributors
All authors participated in the interpretation of study results, and in the drafting, critical revision, and approval of the final version of the manuscript. Y. Sakurai, S. Horii, K. Nakamura, and A. Nishimura were involved in the study concept and design. Y. Sakata was responsible for data collection and was the principal investigator. H. Okamoto performed the statistical analysis.
Conflict of interest
Y. Sakurai, M. Shiino, S. Horii, H. Okamoto, K. Nakamura, and A. Nishimura are employees of Takeda Pharmaceutical Company Limited and have no other relevant disclosures to declare. Y. Sakata has no relevant conflicts of interest to declare.
Ethical Approval
The institutional review board (IRB) for SOUSEIKAI Hakata Clinic was constituted according to the requirements of the participating location. The IRB was responsible for the initial and continuing review and approval of the clinical study in accordance with the requirements of the Japanese Good Clinical Practice (GCP). This study was conducted with respect for the individual participants according to the protocol, the World Medical Association Declaration of Helsinki, the International Conference on Harmonisation (ICH) Harmonised Tripartite Guideline for GCP, and relevant Japanese regulations.
Informed Consent
Before undergoing any study procedures, subjects signed and dated the subject informed consent form, and each subject was given a copy of the form. The written informed consent form was consistent with the elements of informed consent as described in the World Medical Association Declaration of Helsinki and the ICH Harmonised Tripartite Guidelines for GCP
Other Contributors/Acknowledgments
The authors would like to thank all study participants. The authors thank Richard Jenkins, Fiona Steinkamp, Liping Pan, and Siddharth Bhatia, all from Takeda, for reviewing the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sakurai, Y., Shiino, M., Horii, S. et al. Pharmacokinetic Drug–Drug Interactions Between Vonoprazan and Low-Dose Aspirin or Nonsteroidal Anti-inflammatory Drugs: A Phase 2, Open-Label, Study in Healthy Japanese Men. Clin Drug Investig 37, 39–49 (2017). https://doi.org/10.1007/s40261-016-0455-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-016-0455-2