
Abstract
Background and Objective
Metered-dose inhalers require patients to coordinate inhalation with actuation. The present albuterol multi-dose dry-powder inhaler (mDPI) does not require patients to coordinate inspiration with actuation, thereby simplifying delivery of albuterol to the lungs. The aim of the present study was to compare the efficacy, pharmacokinetics, pharmacodynamics, extrapulmonary pharmacodynamics, and safety of albuterol (salbuterol) delivered via a ProAir® hydrofluoroalkane (HFA) metered-dose inhaler and an mDPI.
Methods
Two double-blind, randomized, double-dummy, crossover, multicenter, placebo-controlled studies in persistent asthma patients were conducted. Study 1: 47 adult patients were treated with cumulative doses of albuterol mDPI or ProAir HFA (90 µg/inhalation; 1 + 1 + 2 + 4 + 8 inhalations) or placebo. Study 2: 71 patients aged ≥12 years were randomly assigned to receive 90 or 180 μg of albuterol mDPI or ProAir HFA, or placebo. Primary efficacy endpoints were baseline-adjusted forced expiratory volume in 1 s (FEV1) at 30 min (30-min FEV1) after each cumulative dose (Study 1) and FEV1 area under the effect curve over 6 h (FEV1 AUEC0–6) after dosing (Study 2).
Results
Study 1: differences, with corresponding 90 % confidence intervals, between albuterol mDPI and ProAir HFA in FEV1 after each cumulative dose and in FEV1 AUEC0–6 after the final dose were within pre-established equivalence limits. The difference in FEV1 at high vs. low doses was significant for both active treatments (p < 0.0001). Active treatments were similar in systemic exposure, extrapulmonary pharmacodynamics, and safety. Study 2: mean FEV1 AUEC0–6 was significantly greater than for placebo for both doses of albuterol mDPI and ProAir HFA (p < 0.0001). Albuterol mDPI was comparable to ProAir HFA at 90 and 180 µg. Both study treatments were generally well tolerated.
Conclusion
The bronchodilatory efficacy and pharmacokinetic/pharmacodynamic profiles of albuterol mDPI and ProAir HFA are comparable, with a safety profile consistent with that of inhaled albuterol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Albuterol delivered using a multi-dose dry-powder inhaler (mDPI) has comparable efficacy, safety, pharmacokinetics, and extrapulmonary pharmacodynamics to albuterol delivered using a hydrofluoroalkane metered-dose inhaler. |
An mDPI for delivery of albuterol provides asthma patients a new choice of inhaler device, which might particularly benefit those experiencing difficulty using a conventional metered-dose inhaler. |
1 Introduction
Asthma affects approximately 18.7 million adults in the USA [1] and imposes considerable and persistent burden in terms of morbidity, quality of life, and healthcare expenses [2]. Guidelines recommend the use of short-acting β2-agonists as the treatment of choice for relief of acute asthma symptoms and prevention of exercise-induced bronchospasm [3]. Albuterol (salbuterol) has a well-established safety and efficacy profile, supported by more than 30 years of extensive clinical use as a bronchodilator [4–6]. Inhalation of these medications as an aerosol is the preferred mode of administration, which achieves high drug concentrations in the airways while minimizing the occurrence of systemic side effects [7].
The predominant mode of albuterol administration is via hydrofluoroalkane (HFA) metered-dose inhalers (MDIs). These devices allow direct delivery of the medication to the airways [8], while minimizing oral deposition and systemic exposure (compared with oral therapies), which lead to side effects [9]. However, many patients are unable to properly coordinate actuation of the MDI device with inspiration [9, 10], potentially resulting in compromised drug delivery and suboptimal clinical benefit [11–13].
This problem has been addressed by the development of a novel albuterol multi-dose dry-powder inhaler (mDPI) that delivers the drug as the patient inhales, thus simplifying and improving albuterol delivery to the airways [14, 15].
The results of two studies that compared the efficacy and safety of the new albuterol mDPI and albuterol HFA MDI (ProAir® HFA) in patients with persistent asthma are reported herein. In the cumulative-dose phase I study, the efficacy, pharmacokinetics, extrapulmonary pharmacodynamics, and safety of these two treatments were compared in patients. In the single-dose crossover study, two doses of the albuterol mDPI were compared to corresponding doses of ProAir HFA to confirm they provided similar efficacy and safety. These two studies supported the dose selection of albuterol mDPI for subsequent clinical development.
2 Methods
2.1 Participants
Patients were eligible for inclusion in the studies if they met the following criteria: Study 1: male or female patients aged 18–45 years; Study 2: male or female patients aged ≥12 years. Patients in both studies were included if they were non-smokers for ≥12 months preceding screening and had persistent asthma (according to National Asthma Education and Prevention Program Guidelines) [16] of ≥6 months’ duration that required stable doses of inhaled corticosteroids (ICS; fluticasone propionate ≤500 µg/day or equivalent for 4 weeks preceding screening). All patients had forced expiratory volume in 1 s (FEV1) of 50–80 % of the predicted values for age-, height-, sex-, and race-matched reference values (National Health and Nutrition Examination Survey III) [17] at screening and evidence of reversible bronchoconstriction as verified by a ≥15 % increase in FEV1 within 30 min (30-min FEV1) after inhalation of albuterol 180 µg via MDI. Patients had to have the ability to perform spirometry and peak expiratory flow determinations and be in general good health with a clinically acceptable medical history, physical examination results, and vital signs. All patients had to demonstrate proficiency in use of both the mDPI and HFA MDI to be eligible for the studies and receive their dose during the treatment days.
Major exclusion criteria included known hypersensitivity to albuterol or any of the excipients in the formulations, history of severe milk protein allergy, upper or lower respiratory infection within 14 days (Study 1) or 6 weeks (Study 2) preceding screening, and asthma exacerbation that required oral corticosteroids within the 3 months or hospitalization for asthma within the 6 months preceding screening.
Patients could not have any history of life-threatening asthma. Prohibited concomitant medications included β2-adrenergic receptor agonists, non-selective β-receptor blocking agents such as antihypertensive β-blockers, monoamine oxidase inhibitors, tricyclic antidepressants, systemic corticosteroids, leukotriene modifiers, and, in Study 2, orally inhaled anticholinergics. ICS were continued [no more than 500 µg of fluticasone propionate/day or equivalent dose for another ICS; mean ICS dose at screening (Study 2): 350.4 µg; range: 80–880 µg], short-acting β2-agonists were permitted but had to be withheld 48 h before and during study visits (Study 1) or ≥6 h before spirometry (Study 2), and Study 1 allowed ipratropium use with a washout required 8 h prior to and for the duration of visit assessments.
2.2 Study Designs and Interventions
Study 1: double-blind, randomized, double-dummy, cumulative-dose, two-period crossover, multicenter study (NCT01056159) in 47 adult patients from eight study sites across the USA. This study consisted of a screening visit, followed by a 7- to 14-day run-in period (during which patients were allowed as-needed use of albuterol and ipratropium with specified washouts prior to and during treatment, and, where applicable, were maintained on their dose of ICS), a treatment period with two visits separated by 3–14 days of washout, and a follow-up visit 1–5 days after the second treatment visit. A stable dose of ICS was required for ≥4 weeks preceding the screening visit and during the study. Eligible patients were randomly assigned to receive albuterol mDPI and placebo HFA MDI, or ProAir HFA MDI and placebo mDPI (90 µg/inhalation) in a double-blind, double-dummy, randomized sequence, two-period crossover fashion. Both the treatment sequence and the inhaler sequence (active, placebo) were randomized. After an overnight fast ≥6 h, treatment was administered as 1 + 1 + 2 + 4 + 8 inhalations from each device. Each set of inhalations was separated by 30 min. This resulted in cumulative doses of 90, 180, 360, 720, and 1440 µg for each study treatment. FEV1 was assessed at baseline (the average of measurements taken 30 min and immediately before dose administration) and 30 min after each cumulative dose, with additional hourly assessment up to 6 h after the final dose. In the subset of patients who participated in the pharmacokinetic sub-study, pharmacokinetic assessments continued up to 12 h.
Study 2: randomized, double-blind, double-dummy, placebo-controlled, crossover, multicenter study (NCT01058863) in 71 patients aged ≥12 years from 12 study sites across the USA, consisting of three periods and seven visits. During the 14-day run-in period, regular ICS maintenance asthma treatment was continued. For safety purposes, patients recorded their morning peak expiratory flow in a daily diary (best value of three attempts) and were instructed to contact the investigative site in the case of worsening asthma. Eligible patients were subsequently randomly assigned to receive five single doses of study medication administered in a five-way crossover, double-blind, double-dummy fashion, with a 3- to 7-day washout period between treatment visits. The follow-up period was 3–7 days after the last treatment visit, or at patient discontinuation from the study. Study medication was self-administered at approximately 8.00 a.m. on the day of treatment visits. Each patient was randomly assigned to receive one of five treatments, on separate occasions, via oral inhalation as a single dose (one actuation), including placebo, albuterol mDPI 90 µg, albuterol mDPI 180 µg, ProAir HFA 90 µg, or ProAir HFA 180 µg. On a given treatment day, patients were provided four inhalers, two blinded mDPIs and two blinded HFA MDIs, and self-administered a single dose from each inhaler. At each treatment visit, baseline FEV1 was obtained as the average of two measurements taken 30 min and immediately before dose administration. Post-dose assessments were obtained at 5, 15, 30, and 45 min, and hourly thereafter until 6 h after the final dose.
2.3 Randomization
Eligible patients were randomly assigned via an interactive voice response system to one of four treatment sequences composed of the two possible treatments and the two possible sequences of devices within each treatment period (Study 1). Study 2 used an interactive voice response system to randomly assign eligible patients to one of 10 treatment sequences, and to one of two inhaler sequences.
2.4 Outcomes
2.4.1 Efficacy Assessments
For Study 1, the primary efficacy endpoint was the baseline-adjusted 30-min FEV1 after each of five cumulative doses. The secondary efficacy endpoint was the FEV1 area under the effect curve over 6 h (FEV1 AUEC0–6) after completion of dosing. Additionally, to evaluate a dose response to the escalating doses, a comparison between the FEV1 at the lowest and highest cumulative dose with each device was made. For Study 2, the primary efficacy endpoint was the baseline-adjusted FEV1 AUEC0–6. The secondary endpoint was the baseline-adjusted AUEC0–6 of percent-predicted FEV1 (PPFEV1 AUEC0–6). Other efficacy endpoints included baseline-adjusted maximum FEV1 within 6 h after dosing, time to achievement of and percentage of patients achieving a ≥15 % and a ≥12 % increase from baseline FEV1 within 30 min after dosing, duration of ≥15 and ≥12 % increases in FEV1, and time to maximum FEV1 (time to peak).
2.4.2 Tolerability Assessments
Safety in both studies was assessed via physical examination, vital sign assessment, and monitoring for any spontaneous and elicited adverse events (AEs). In addition, patients in Study 1 underwent electrocardiography and laboratory evaluations.
2.4.3 Pharmacokinetic Assessments
For Study 1 in a subset of patients, the plasma albuterol drug concentration was measured at 15 min after each of the first four cumulative doses and at 15 and 30 min and 1, 2, 3, 4, 6, 8, 10, and 12 h after the final cumulative dose. Pharmacokinetic endpoints included area under the plasma concentration time curve extrapolated to last measurable concentration (AUC0–t), maximum post-dose plasma concentration (C max), and time to observed peak plasma concentration (T max).
2.4.4 Pharmacodynamic Assessments
For Study 1, vital signs (blood pressure and heart rate), electrocardiography (corrected QT intervals), plasma glucose levels, and potassium levels were all assessed at 15 min after each cumulative dose and serially after the final cumulative dose at 0.25, 0.5, 1, 2, 3, and 4 h, with additional assessments of vital signs at 5 and 6 h after the final cumulative dose.
2.5 Statistical Analysis
Study 1: based upon a simulation using results from the IVAX study (IX-100-076), a sample size of 40 patients was found to provide a power of 91 % to accept the hypothesis that the absolute difference between albuterol mDPI and ProAir HFA was <0.2 L for all of the cumulative doses. This would be demonstrated at the 0.05 level of significance if all of the individual confidence bounds for the treatment difference were within the pre-established limits of −0.2, 0.2 L. Approximately 48 patients were to be randomly assigned to ensure that a sample size of 40 was maintained. For the pharmacokinetic sub-study, a sample size of 22 patients was required to provide a power of >90 % to demonstrate that exposure to albuterol mDPI was no greater than with ProAir HFA (one-sided equivalence, using the standard upper equivalence limit of 1.25). Approximately 24 patients were randomly assigned to ensure that 22 patients completed the pharmacokinetic sub-study.
Study 2: comparison of each treatment to placebo was performed to determine an initial level of significance. If significance was not obtained, no further tests were performed. This sequential process assured that the overall alpha level for the entire series was not greater than 0.05. A sample size of 60 patients was required to provide a power of 90 % to detect a difference in baseline-adjusted FEV1 AUEC0–6 of 0.50 L h between active treatment and placebo, using 0.82 L h as the value of the intra-subject standard deviation in the crossover model in a two-sided test at the 0.05 level of significance. Sixty-six patients were to be randomly assigned to ensure an adequate sample size.
The safety population included all randomized patients who received one or more doses of randomized study medication. The per-protocol (PP) population (the primary population for efficacy and pharmacodynamic evaluations in Study 1) included patients who completed the study without any major protocol violations. The pharmacokinetic analysis population (Study 1 only) included all patients randomly assigned in the pharmacokinetic sub-study with valid data at blinded review of assay results before analysis. The intent-to-treat (ITT) population (primary population for efficacy evaluations in Study 2) included all randomly assigned patients who received one or more doses of randomized study medication and had one or more post-baseline assessments. Baseline was considered the pretreatment assessment on each study treatment day; baseline FEV1 was the average of the two pre-dose determinations. Demographic and baseline characteristics, FEV1 changes from baseline, pharmacokinetic and pharmacodynamic data, and AEs were summarized using descriptive statistics.
2.5.1 Efficacy Analyses
For Study 1, the baseline-adjusted 30-min FEV1 after each of the five cumulative doses and the difference in the change from baseline 30-min FEV1 after the last (1440 μg) vs. first (90 μg) dose were analyzed using a mixed-effect analysis of covariance (ANCOVA) model with fixed effects of baseline FEV1, sequence, treatment group, period, and study site, cumulative dose, treatment × cumulative dose, random effect for patient within sequence, and an AR(1) correlation structure between repeated FEV1 measurements. The primary efficacy analysis for Study 1 was based on a comparison of albuterol mDPI and ProAir HFA with regard to the change from baseline in FEV1 obtained at 30 min after dosing following each of the cumulative doses. Study treatments were considered comparable with respect to FEV1 if, at each cumulative dose, the 90 % confidence interval (CI) for the mean between-group difference was within the limits of ±0.20 L. The FEV1 AUEC0–6 was also analyzed with a mixed analysis of variance model with fixed effects of sequence, treatment group, period, and study site, and a random effect for patient within sequence. The two treatments were considered comparable with respect to FEV1 AUEC0–6 after the final cumulative dose if 90 % CIs for the treatment difference were within the limits of ±1.2 L h.
The comparison of interest for Study 2 was the mean difference between each active group and placebo at each dose level. This was performed with a mixed-effect ANCOVA model with fixed effects of sequence, treatment group, period, and study site, and random effect for subject within sequence. Sequential testing ensured that the overall α level for the entire series of tests was ≤0.05. Percentages of patients achieving ≥15 and ≥12 % increases from baseline in 30-min FEV1 for each active treatment and placebo were analyzed with a mixed logistic regression model containing fixed effects for treatment and period and a random term for patient. An exploratory analysis was undertaken to compare the two active treatment estimated means, standard errors, and CIs derived from a linear mixed model with fixed effects of treatment, sequence, period, pooled center, and baseline values and a random effect of patient.
2.5.2 Pharmacokinetic Analyses
In Study 1, plasma albuterol was compared after a cumulative dose of 1440 μg administered by mDPI and by ProAir HFA. Albuterol AUC0–t and C max per treatment were compared using an ANCOVA model with fixed effects of sequence, period, and treatment, and a random effect for patient within sequence, based on log-transformed data. The geometric mean ratio (GMR) between the two study treatments was estimated, and exposure was considered equivalent if the 90 % CIs were between 0.8 and 1.25. The Wilcoxon signed rank test was used for nonparametric comparisons of T max between study treatments.
2.5.3 Pharmacodynamic Analyses
For analysis of pharmacodynamic measures in Study 1, the same model as described for serial FEV1 measures was used. The model was used to construct 90 % CIs for mean treatment differences and 95 % CIs for treatment means to evaluate the comparability of the study treatments; however, formal equivalence limits were not set.
3 Results
3.1 Study Population
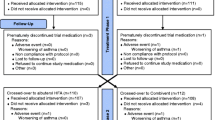
Patient disposition is shown in Fig. 1. After unblinding in Study 1, significant data anomalies at a single study site resulted in two additional analysis populations being defined (the primary PP and pharmacokinetic PP populations), which excluded pharmacokinetic, efficacy, and pharmacodynamic data from this study site. As a result, the primary PP and pharmacokinetic PP populations were composed of 39 and 16 patients, respectively. Patient demographics at baseline for studies 1 and 2 are summarized in Table 1.

Patient disposition in Study 1 and Study 2. ITT intent to treat, PK pharmacokinetics, PP per protocol. aAll patients from one investigational site (n = 8) were excluded because of uncertainty regarding albuterol exposure. bNinety-one patients did not qualify for the study based on reversibility or spirometry criteria (study-qualifying FEV1 was not within 50–80 %, or nonspirometry inclusion/exclusion). cOne subject was incorrectly entered as “randomized” and was withdrawn from the study before receiving study treatment. dPK data were excluded from two patients in this population
3.2 Study 1 Results
3.2.1 Efficacy
In the primary PP population, albuterol mDPI and ProAir HFA both were associated with increases ranging from 0.42 to 0.72 L in baseline-adjusted 30-min FEV1 after each of the five cumulative doses (Table 2). Between-treatment differences ranging from 0.03 to 0.07 L were observed, and the 90 % CIs were within the pre-defined limits of ±0.20 L (Table 2), indicating that the two study treatments were comparable.
For the secondary endpoint (baseline-adjusted FEV1 AUEC0–6 after the final cumulative dose), with 90 % CIs being contained within the pre-defined limits of 1.2 L h, statistically significant dose responses (both p < 0.0001) were observed for each product in that the differences in change from baseline in 30-min FEV1 post-dose after 1440 vs. 90 μg were 0.269 ± 0.029 and 0.246 ± 0.029 L for albuterol mDPI and ProAir HFA, respectively. Peak changes in FEV1 were modestly larger with ProAir HFA than with albuterol mDPI (Fig. 2).

Least-square mean change from baseline in FEV1 at each dose and following the cumulative dose in the PP population. FEV 1 forced expiratory volume in 1 s, PP per protocol population. Bars indicate standard error of the mean. Arrows (↑) indicate dosing times at 0, 0.5, 1, 1.5, and 2 h
Findings of the pharmacokinetic sub-study (primary pharmacokinetic PP population) indicated that the systemic exposure (AUC0−t) after albuterol mDPI (23,277 pg h/mL) was comparable to ProAir HFA (20,939 pg h/mL), with the GMR 1.109 (90 % CI, 1.04, 1.19) and the 90 % CI contained within the equivalence range of 0.8–1.25. In contrast, C max was numerically higher with albuterol mDPI (4422.8 vs. 3303.8 pg/mL), with the GMR 1.339 pg/mL (90 % CI 1.17, 1.53). The mean T max was numerically earlier with albuterol mDPI (2.48 vs. 3.05 h).
No clinically meaningful changes in extrapulmonary pharmacodynamic effects were seen with either treatment, including plasma glucose level, plasma potassium level, corrected QT intervals, blood pressure, and heart rate (data not shown). The changes in these measures were comparable between albuterol mDPI and ProAir HFA.
3.2.2 Safety
Albuterol mDPI and ProAir HFA were well tolerated and had comparable AE profiles. The most frequently reported treatment-emergent AEs were tremor (17.4 vs. 13.0 %), headache (8.7 vs. 4.3 %), and palpitations (8.7 vs. 2.2 %); the majority of these events were reported at the cumulative doses of 720 and 1440 μg. No deaths, serious AEs, or discontinuations owing to an AE were reported.
3.3 Study 2 Results
3.3.1 Efficacy
Baseline FEV1 values were similar for each of the five treatment arms, ranging between 2.15 and 2.17 L. Baseline-adjusted FEV1 values over a 6-h period after dosing are shown in Fig. 3a. The difference (standard error) in mean baseline-adjusted FEV1 AUEC0–6 from placebo for each active treatment was statistically significant [albuterol mDPI 180 µg, 1.15 (0.136); albuterol mDPI 90 µg, 0.97 (0.136); ProAir HFA 180 µg, 1.08 (0.136); ProAir HFA 90 µg, 0.88 (0.136); all p < 0.0001].

Baseline-adjusted mean a FEV1 and b PPFEV1 up to 6 h after dosing by dose level and device type in Study 2 (ITT population). FEV 1 forced expiratory volume in 1 s, HFA hydrofluoroalkane, ITT intent to treat, PPFEV 1 percent-predicted forced expiratory volume in 1 s at 5, 15, 30, and 45 min and 1, 2, 3, 4, 5, and 6 h. Bars indicate standard error of the mean. Arrow (↑) indicates dosing at time 0
Pre-treatment values for PPFEV1 at baseline were similar for each of the five treatment arms, ranging between 64.89 and 65.58 %. Baseline-adjusted PPFEV1 values over a 6-h period after dosing are shown in Fig. 3b; all active treatments showed improvement over the 6-h post-dose period (Table 3). For this secondary endpoint, the estimated mean PPFEV1 AUEC0–6 values for each individual dose of albuterol mDPI and ProAir HFA were statistically significantly greater than for placebo (all p < 0.0001; Table 3). The mean baseline-adjusted maximal FEV1 value over a 6-h post-dose period was statistically significantly greater for all active treatments compared with that of placebo (all p < 0.0001).
An exploratory analysis found that albuterol mDPI and ProAir HFA were not significantly different at both the 90- and 180-µg dosages, as the 90 % CIs for treatment differences in estimated mean FEV1 AUEC0–6 values included zero. The analysis indicated that the magnitude of the differences was quite small (Table 4). An additional analysis explored the dose-response relationship by comparing the difference between the two doses for each inhaler. Although there was a numerical improvement in response at the higher dose for both inhalers, differences between doses in FEV1 AUEC0–6 were not statistically different for either inhaler (Fig. 4).

Mean baseline-adjusted FEV1 AUEC0–6 (L h) after dosing by dose level and device type for active treatment groups in Study 2 (ITT population). AUEC 0–6 area under the effect curve over a period of 6 h, FEV 1 forced expiratory volume in 1 s, HFA hydrofluoroalkane, ITT intent-to-treat, mDPI multi-dose dry-powder inhaler
The mean overall times to maximum increase from baseline in FEV1 after treatment with albuterol mDPI, ProAir HFA, and placebo are summarized in Table 3. Statistically significantly higher percentages of patients achieved ≥15 or ≥12 % increases in 30-min FEV1 from baseline with all active treatments compared with placebo (all p < 0.0001; Table 3). The mean overall times to ≥15 and ≥12 % increases in FEV1 from baseline with active treatment were similar across all active treatments (Table 3). Mean overall durations of ≥15 and ≥12 % increases in FEV1 from baseline were also similar across all active treatments (Table 3).
3.3.2 Safety
No deaths, serious AEs, or discontinuations were reported during the study. The most common types of AEs were upper respiratory tract infection (n = 1; albuterol mDPI 90 µg), viral infection (n = 1; placebo), localized infection (n = 1; ProAir HFA 180 µg), moderate hypersensitivity (n = 1; placebo), and food allergy (n = 1; albuterol mDPI 90 µg) (Table 5). No occurrences of paradoxical bronchospasm or asthma exacerbation were reported during the study.
4 Discussion
The novel dry-powder albuterol mDPI showed comparable overall efficacy, pharmacokinetics, pharmacodynamics, and safety compared with the conventional MDI ProAir HFA.
Robust increases in baseline-adjusted FEV1 at each cumulative dose level were seen with both albuterol mDPI and ProAir HFA; these improvements met the pre-defined criteria for comparability between the two treatments (Study 1). Bioequivalence of the two study treatments was demonstrated with regard to systemic exposure (AUC0–t) in the pharmacokinetic sub-study, although numerically higher C max and lower T max values were seen with albuterol mDPI. The small sample size (16 patients included in the primary pharmacokinetic PP population) might have contributed to the differences in C max and T max, but the comparable changes in extrapulmonary pharmacodynamic measures between albuterol mDPI and ProAir HFA at and above therapeutic doses indicated that these differences were not clinically meaningful. However, given the sample size, the results are considered preliminary.
Although a higher incidence of treatment-emergent AEs was reported with albuterol mDPI at supratherapeutic doses (720 and 1440 μg), treatments were generally well tolerated at normally prescribed dosages.
Study 2 further evaluated the comparability of albuterol mDPI and ProAir HFA inhalers at doses of 90 and 180 µg in adolescent and adult patients with persistent asthma. Albuterol mDPI and ProAir HFA both demonstrated greater baseline-adjusted efficacy outcomes than placebo, as shown by statistically significant improvement in FEV1 AUEC0–6, PPFEV1 AUEC0–6, and maximum FEV1 over a period of 6 h. In addition, ≥60 and ≥44 % of patients receiving active treatment exhibited increases from baseline in 30-min FEV1 of ≥12 and ≥15 %, respectively.
Although not powered to detect equivalence between the mDPI and HFA MDI devices, exploratory analyses showed that bronchodilator efficacy with albuterol mDPI was comparable to that of ProAir HFA at equivalent doses. Albuterol mDPI and ProAir HFA both were well tolerated at 90 and 180 µg, with safety profiles consistent with the well-established safety profile of albuterol. Specifically, there were no meaningful differences in the incidence of AEs or post-dose changes in vital signs for albuterol mDPI compared with placebo or equivalent doses of ProAir HFA. There were no reports of paradoxical bronchospasm or asthma exacerbation. Overall, the incidence of AEs was low.
Drug delivery via a device that does not require the coordination of actuation and inhalation might be a worthwhile consideration for any patient, but might particularly benefit those who have difficulty using conventional MDIs or who are otherwise unable to use MDI devices optimally. In adults and children who have poor inhaler technique, pressurized MDIs that do not require coordination of actuation and inhalation have been associated with enhanced drug deposition, improved drug delivery, and improved bronchodilator response, which might result in more favorable clinical outcomes compared with conventional MDIs [18]. Additionally, patients and healthcare professionals both report higher levels of satisfaction and a reduction in administration errors with the use of breath-actuated devices [19]. An mDPI inhaler requires a lesser degree of coordination with the device delivery because it is the patient’s inspiratory effort that governs the rate of drug delivery throughout the entire inhalation. Availability of a multi-dose dry-powder inhaler for albuterol would provide patients and their healthcare professionals with an alternative choice of inhaler device.
Additional limitations of using an MDI apply to all currently marketed inhalers and are related to maintenance and administration, which also are considerably improved with the mDPI. All albuterol MDIs require cleaning with water on a regular basis and shaking, priming, or re-priming before use. Patients need to actuate and inhale the drug quickly before the content settles, and inhaler priming and re-priming account for wasted drug product. Failure to adhere to these requirements for albuterol HFA MDI can impact the drug delivery and have clinical consequences [10–13]. With the mDPI, regular cleaning (other than mouthpiece with a dry cloth if needed), shaking, priming, and re-priming are not needed. This further simplifies the use of the mDPI for patients and helps to ensure drug delivery and clinical benefits.
Limitations of these two studies include the single-dose design, the small patient numbers (pharmacokinetic/pharmacodynamic measurements), and the lack of patient-preference questionnaires to assess inhaler preferences. Furthermore, the technique-related advantages of the mDPI could not be studied because patients incapable of using an MDI properly (about half the patients in real-world studies) were excluded from this study [9, 10]. The strengths of these studies include the crossover design, the double-blind, double-dummy dosing, and the high rate of study completion achieved (≥95 % in both).
5 Conclusions
The results support the use of a novel dry-powder albuterol mDPI for the management of acute asthma symptoms in patients with persistent asthma. Efficacy outcomes with albuterol mDPI were improved compared with placebo and comparable to those of the conventional ProAir HFA. Availability of a breath-activated device for delivery of albuterol that is easy to maintain and use would provide healthcare professionals and patients with more options for rescue bronchodilator therapy.
References
Centers for Disease Control and Prevention. FastStats: asthma. Available at: http://www.cdc.gov/nchs/fastats/asthma.htm. Accessed 13 Aug 2015.
Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the national asthma education and prevention program guidelines. Am J Respir Crit Care Med. 2002;166:1044–9.
Expert Panel Report 3 (EPR-3). Guidelines for the diagnosis and management of asthma: summary report 2007. J Allergy Clin Immunol. 2007;120:S94–138.
Proair HFA [package insert]. Horsham, PA: Teva Respiratory, LLC; 2012.
Bronsky E, Bucholtz GA, Busse WW, et al. Comparison of inhaled albuterol powder and aerosol in asthma. J Allergy Clin Immunol. 1987;79:741–7.
Kemp JP, Furukawa CT, Bronsky EA, et al. Albuterol treatment for children with asthma: a comparison of inhaled powder and aerosol. J Allergy Clin Immunol. 1989;83:697–702.
Dolovich MB, Ahrens RC, Hess DR, et al. Device selection and outcomes of aerosol therapy: evidence-based guidelines: American College of Chest Physicians/American College of Asthma, Allergy, and Immunology. Chest. 2005;127:335–71.
Given J, Taveras H, Iverson H, et al. Prospective, open-label assessment of albuterol sulfate hydrofluoroalkane metered-dose inhaler with new integrated dose counter. Allergy Asthma Proc. 2013;34:42–51.
Cochrane MG, Bala MV, Downs KE, et al. Inhaled corticosteroids for asthma therapy: patient compliance, devices, and inhalation technique. Chest. 2000;117:542–50.
Crompton GK, Barnes PJ, Broeders M, et al. The need to improve inhalation technique in Europe: a report from the Aerosol Drug Management Improvement Team. Respir Med. 2006;100:1479–94.
Lindgren S, Bake B, Larsson S. Clinical consequences of inadequate inhalation technique in asthma therapy. Eur J Respir Dis. 1987;70:93–8.
Chrystyn H, Price D. Not all asthma inhalers are the same: factors to consider when prescribing an inhaler. Prim Care Respir J. 2009;18:243–9.
Giraud V, Roche N. Misuse of corticosteroid metered-dose inhaler is associated with decreased asthma stability. Eur Respir J. 2002;19:246–51.
Scichilone N, Benfante A, Bocchino M, et al. Which factors affect the choice of the inhaler in chronic obstructive respiratory diseases? Pulm Pharmacol Ther. 2015;31:63–7.
Demoly P, Hagedoorn P, de Boer AH, et al. The clinical relevance of dry powder inhaler performance for drug delivery. Respir Med. 2014;108:1195–203.
Expert Panel Report 3 (EPR3). Guidelines for the diagnosis and management of asthma. Bethesda, MD: National Heart, Lung, and Blood Institute, US Department of Health and Human Services. 2007. Available at: http://www.nhlbi.nih.gov/guidelines/asthma/asthgdln.htm. Accessed on 19 Aug 2015.
Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159:179–87.
Newman SP, Weisz AW, Talaee N, et al. Improvement of drug delivery with a breath actuated pressurised aerosol for patients with poor inhaler technique. Thorax. 1991;46:712–6.
Price DB, Pearce L, Powell SR, et al. Handling and acceptability of the Easi-Breathe device compared with a conventional metered dose inhaler by patients and practice nurses. Int J Clin Pract. 1999;53:31–6.
Acknowledgments
The authors acknowledge and thank the investigators from Study 1 and Study 2 and their respective teams for their valuable contributions during the conduct of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Teva Branded Pharmaceutical Products R&D, Inc. Medical writing and editorial assistance during the preparation of this manuscript was provided by ApotheCom, with funding provided by Teva.
Conflicts of interest
Dr. Kerwin has served on advisory boards for Amphastar, AstraZeneca (Pearl), Mylan, Novartis, Sunovion, and Theravance; participated on speaker panels for Amphastar, AstraZeneca (Pearl), Boehringer Ingelheim, Forest, Mylan, Novartis, Sunovion, and Theravance; and has received travel reimbursement for presentations at meetings from Forest, Novartis, and Teva. He has conducted multicenter clinical research trials for approximately 40 pharmaceutical companies. Dr. Miller receives research funding from Teva and served as principal investigator for Teva through Northeast Medical Research Associates. Drs. Iverson, Shah, Taveras, and Wayne are employed by Teva Branded Pharmaceutical Products R&D and may hold stock or stock options in Teva. Dr. Lepore was employed by Teva Branded Pharmaceutical Products R&D at the time the study was conducted and holds stock or stock options in Lupin Inc.
Ethical approval
All procedures in both studies were conducted in accordance with the ethical principles based on the 1964 Declaration of Helsinki (and its amendments) and Good Clinical Practice. Both studies were approved by the institutional review board at each study site.
Informed consent
All patients from both studies provided written informed consent before screening.
Rights and permissions
About this article

Cite this article
Kerwin, E.M., Taveras, H., Iverson, H. et al. Pharmacokinetics, Pharmacodynamics, Efficacy, and Safety of Albuterol (Salbuterol) Multi-dose Dry-Powder Inhaler and ProAir® Hydrofluoroalkane for the Treatment of Persistent Asthma: Results of Two Randomized Double-Blind Studies. Clin Drug Investig 36, 55–65 (2016). https://doi.org/10.1007/s40261-015-0346-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-015-0346-y