
Abstract
Age-related macular degeneration (AMD) is a leading cause of visual impairment and blindness in older adults. The prognosis for the neovascular type of advanced AMD improved with the introduction of biological drugs with antiangiogenic properties, beginning with off-label bevacizumab, which was first used intravitreally in 2006. These drugs target newly formed vessels that grow beneath the center of the retina, causing loss of central vision, and they can help to maintain or improve vision. Repeated intravitreal injections are needed to achieve prolonged inhibition of proangiogenic cytokines, primarily vascular endothelial growth factor (VEGF). Major regulatory agencies have approved several molecules for AMD treatment, including ranibizumab, aflibercept, and brolucizumab. The development of further drugs was mainly targeted at prolonging anti-VEGF inhibition—thus reducing the frequency of injections—and expanding the biological targets of proangiogenic cytokine inhibition. Finally, biosimilars are already being marketed in some countries, allowing the containment of costs of AMD treatment, which are growing steadily in many settings because of the need for long-term treatment. This review summarizes the properties and clinical profiles of anti-VEGF biological drugs that are approved to treat neovascular AMD as well as ongoing research on molecules that may be marketed in the near future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Age-related macular degeneration (AMD) is one of the most common causes of vision loss in older adults. The prognosis of the neovascular form of advanced AMD has improved since the introduction of biological drugs with antiangiogenic properties. |
Several molecules that aim to bind vascular endothelial growth factor (VEGF) have been approved for AMD treatment, including ranibizumab, aflibercept, and brolucizumab. |
Newer drugs are mainly targeted at prolonging anti-VEGF inhibition to reduce the frequency of injections and at expanding the biological targets of proangiogenic cytokine inhibition. |
1 Etiology, Pathogenesis, Epidemiology, and Clinical Outcomes of Age-Related Macular Degeneration (AMD)
Age‐related macular degeneration (AMD) affects the central part of the retina, causing progressive loss of central vision [1, 2]. The early stages of AMD are often asymptomatic, and people can be diagnosed with macular drusen during even routine eye examinations. Drusen are due to the accumulation of lipid material, which accumulates in deposits underneath the retinal pigment epithelium (RPE). In advanced AMD stages, the RPE may atrophy completely, first in small focal areas, then sometimes progressing to widespread, or geographic, atrophy. Late AMD can also be exudative, or “wet,” when newly formed blood vessels grow under the RPE and, occasionally, into the subretinal space (neovascular AMD [nAMD]). This neovascular tissue (choroidal neovascularization [CNV]) causes subretinal hemorrhage and intraretinal fluid accumulation, which often results in increased scarring of the retina with disruption of the macular tissues. Retinal damage can cause the development of a blind spot in central vision, which initially affects tasks such as reading and face recognition, and can progress to a level that limits mobility and personal autonomy. Once a patient develops nAMD in one eye, the fellow eye is at high risk of developing the same condition within 5 years, also depending on fundus lesions and age [3].
On a global scale, AMD and diabetic retinopathy are the fourth and fifth causes of blindness after cataract, glaucoma, and refractive error. Population aging and growth saw cases of blindness rise by 50–60% between 1990 and 2020, although their age-adjusted prevalence decreased by about 30%. Clearly, aging is the greatest risk factor for AMD. Recent genetic research focusing on polymorphisms showed that genetic risk profiles in East Asian populations resembled those in Europeans and were predicted to result in a large increase in the incidence of AMD over the coming years as compared with Chinese populations [4, 5].
From a clinical perspective, AMD is classified into stages such as early, intermediate, and late based on color fundus photography. With rapid advances in ophthalmic imaging, including the increasing resolution and software processing achieved with optical coherence tomography (OCT) and OCT angiography, the fundus photographic classification has evolved towards an integrated classification based on multimodal imaging of the chorioretinal structure [6]. A systematic review of systematic reviews showed that visual impairment affects quality of life and that numerous interventions, primarily cataract surgery and intravitreal anti-vascular endothelial growth factor (anti-VEGF) injections, have a measurable beneficial impact on quality of life [7].
Over the last decade, the treatment of macular disease has been improved with the advent of anti-VEGF drugs such as pegaptanib, ranibizumab, aflibercept, and bevacizumab used off label. In addition, glucocorticoids were also used as therapy for retinal diseases such as diabetic macular edema or retinal vein occlusion. Dexamethasone or fluocinolone implants have the advantage of the longest surgery-free period (not less than 6 months) because of their formulation.
Pegaptanib was the first anti-VEGF approved in AMD but has been completely replaced with the advent of the other drugs. Moreover, different molecules are still under investigation to improve the results already achieved, including biosimilars and drug-delivery systems, as described in detail in Sects. 2.3 and 3.
When reporting evidence for intravitreal anti-VEGF treatments for ocular disease, an important premise is that drug type and treatment regimen are equally key components of treatment effectiveness. Randomized controlled trials (RCTs) typically standardize the treatment regimen to compare drug efficacy, but some RCTs compared different regimens of the same drug. A systematic review showed that less-than-monthly regimens use fewer injections than monthly regimens but have slightly poorer visual function outcomes [8]. Moreover, as-needed (pro re nata [PRN]) regimens, in which injections are delivered when CNV recurs, may be slightly less effective than treat and extend (T&E) regimens, in which CNV recurrence is managed with progressively larger treatment intervals and injections are delivered regardless of whether new recurrence is observed. Unfortunately, a variable and sometimes large gap exists between clinical practice and RCTs, since patients are often undertreated in many clinical settings and any visual benefit with anti-VEGF injections for AMD may be smaller or not maintained in the long term.
This evidence allows us to make a few statements that support the conduct of this review. First, vision impairment is an expanding global health issue that affects quality of life. Second, preventive strategies and therapeutic interventions exist to maintain or restore quality of life in individuals with vision impairment worldwide. Third, geographic differences exist regarding the leading causes of vision impairment. The complications of the most common retinal vascular diseases—diabetic retinopathy and AMD—are treated with intravitreal antiangiogenic therapy using anti-VEGF drugs, of which several molecules exist or are being developed, including biosimilars and drug-delivery systems. Given the huge public health impact in terms of health benefits and use of human and drug resources, the aim of this review was to summarize the evidence on anti-VEGF drugs for treatment of AMD, including their strengths and limitations.
2 Anti-Vascular Endothelial Growth Factor (VEGF) Drugs
2.1 Approved for Neovascular AMD
2.1.1 Pegaptanib
Pegaptanib sodium was the first anti-VEGF drug approved for intravitreal use in the treatment of nAMD in humans [9, 10]. The molecular structure is an RNA aptamer (polyethylene glycol-linked molecule) that binds the VEGF165 and larger isoforms, sequestering VEGF165 and therefore preventing it from activating its receptor. Pegaptanib sodium was approved for the treatment of nAMD in 2004 on the strength of the results of the phase III VISION trial [11] (see Table 1 for the full names of trials cited in this review). However, its poorer efficacy compared with other available anti-VEGF drugs means pegaptanib is no longer recommended for treatment and has been completely replaced.
2.1.2 Ranibizumab
2.1.2.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
Ranibizumab (Lucentis®; Novartis, Basel, Switzerland; Genentech; South San Francisco, CA, USA) is a 48-kDa recombinant humanized immunoglobulin (Ig)-G1k isotype monoclonal antibody fragment (Fab) devoid of the Fc portion. Ranibizumab binds all VEGF-A isoforms.
The vitreous half-life (t½) was 7.2–9 days, and ranibizumab appeared to reach maximum serum concentration approximately 0.5 days after intravitreal administration that was 90,000 times lower than those in the vitreous. The absence of the Fc region makes the molecule susceptible to systemic metabolism, and its serum t½ was estimated at 2 h [10].
Previous animal studies reported a vitreous t1/2 of 2.88 days after a 0.5 mg/0.05 mL single dose in rabbit, and no serum concentration was detected [12]; studies in two different monkey models reported a vitreous t1/2 of 2.63 and 2.73 days, respectively [13, 14] (Table 2).
2.1.2.2 Efficacy and Registration Studies
Several clinical trials were conducted to test the efficacy and safety of ranibizumab in the treatment of nAMD.
MARINA was a 2-year phase III multicenter study in minimally classic/occult neovascularization to evaluate the efficacy and safety of ranibizumab 0.3 and 0.5 mg compared with sham injections. Best corrected visual acuity (BCVA) significantly improved in the ranibizumab groups compared with controls (p < 0.001 for both dosage groups) at month 12 [20, 21].
ANCHOR was an RCT that included classic CNV compared with verteporfin photodynamic therapy (PDT). A clinical benefit in terms of improvement (BCVA) was reported at the 1- and 2-year follow-up (FU) visits [22, 23].
HORIZON was an extension of MARINA and ANCHOR that assessed ranibizumab treatment for 2 additional years and suggested good tolerance for a period ≥ 4 years, although an incremental decline in BCVA improvement was reported [24].
Patients enrolled in these last three studies were also analyzed in SEVEN-UP, a long-term multicenter cohort study that revealed that one-third of patients had poor outcomes, with a visual decline of ≥ 15 letters at year 7 [24, 25].
PIER was another multicenter sham-controlled RCT that tested ranibizumab 0.3 and 0.5 mg. Patients with nAMD were treated quarterly after three monthly loading doses and reported a significant improvement in BCVA at months 12 and 24 (p < 0.0001) [26, 27].
Subsequently, HARBOR investigated the efficacy and safety of ranibizumab 0.5 or 2.0 mg administered monthly or PRN in patients with subfoveal nAMD and reported that the PRN regimen and a dose of 0.5 mg resulted in better visual outcomes than monthly ≥ 0.5 mg doses of ranibizumab at month 24 [28].
More recently, T&E was proposed as an alternative strategy to gain similar BCVA results with a significantly lower number of injections than the monthly regimen [29]. These results were also confirmed in CANTREAT at 12 and 24 months (Table 3).
2.1.3 Bevacizumab
2.1.3.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
Bevacizumab is a fully humanized IgG1 containing the Fc portion; it binds all VEGF-A isoforms and has a molecular weight (Mw) of 148 kDa.
The first indication for intravenous bevacizumab was in metastatic colon cancer; the intravitreal formulation was administered “off label” to treat nAMD and diabetic macular edema (at a much lower dose of 1.25 mg/0.05 mL) [1]. In Italy, it was included in a register of drugs approved for off-label use.
Human studies demonstrated a vitreous t½ range of 2.5–7.3 days (mean 4.9 days) following a 1.25 mg/0.05 mL single dose [15]. Other studies reported a serum t½ of 18.7 days after three single intravitreal doses, with a serum concentration of 1.58 nM, which is higher than the half-maximal inhibition for VEGF inhibitors of 0.668 nM [16, 17, 36].
Animal studies demonstrated a vitreous t½ of 4.32–5.95 days in different rabbit models [37, 38] and a higher concentration in the aqueous humor of the fellow eye than in the vitreous [37]. These results suggested possible systemic circulation through the anterior route, reaching the aqueous and then the vitreous [37].
2.1.3.2 Efficacy and Registration Studies
The ABC trial was the first prospective multicenter RCT in the UK to compare bevacizumab 1.25 mg (three loading doses with further treatment as needed at 6-week intervals) with standard of care (PDT for predominantly classic CNV; pegaptanib or sham for occult or minimally classic CNV) and reported the superiority of this anti-VEGF drug with improved BCVA on average at week 54 [33].
In CATT, which compared the efficacy and safety of bevacizumab and ranibizumab, bevacizumab was noninferior to ranibizumab in monthly and PRN regimens at 1 and 2 years [30, 31].
Data from a long-term post hoc analysis showed that the visual gain was not maintained at 5 years, although one-half of patients had 20/40 or better BCVA [39].
The IVAN study was a multicenter randomized noninferiority study that compared the efficacy and safety of bevacizumab 1.25 mg and ranibizumab 0.5 mg in patients with nAMD. Enrolled patients were treated with monthly or PRN injections, with a monthly review [39]. Results from the comparison of these drugs were inconclusive, as bevacizumab was neither inferior nor equivalent to ranibizumab in either regimen. The results of other comparative studies such as the BRAMD study, the French GEFAL study, and the Australian MANTA study supported the noninferiority of bevacizumab in comparison with ranibizumab [32, 39,40,41,42].
These results confirmed that the use of anti-VEGF was the major long-term therapeutic choice for nAMD [39] (Table 3).
2.1.4 Aflibercept
2.1.4.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
Aflibercept, or VEGF Trap-Eye (EYLEA®; Bayer HealthCare, Berlin, Germany; Regeneron Pharmaceuticals Inc.; Tarrytown, NY, USA), is a 115-kDa recombinant (r-fusion) protein obtained by combining the Fc region of IgG1 with two binding domains of VEGF receptor type 1 and type 2 [1, 10]. Aflibercept traps all isoforms of VEGF-A, VEGF-B, and placental growth factor (PlGF) [10, 43].
Using a mathematical model, a vitreous t½ of 7.13 days was estimated [10]. The serum t½ for aflibercept was 11.4 days following 3-monthly intravitreal injections of 2 mg/0.05 mL [16, 17]. The peak was achieved within 1–3 days post-dose, and the plasma concentration quickly decreased below the lower limit of quantification [10, 44].
Animal studies showed a vitreous t½ of 4.58 and 2.44 days in rabbit and monkey models, respectively [13, 45] (Table 2).
2.1.4.2 Efficacy and Registration Studies
VIEW 1 and 2 were phase III RCTs evaluating different dose regimens (0.5 mg monthly vs. 2 mg monthly or 2 mg bimonthly after 3 once-monthly loading doses) of aflibercept compared with ranibizumab 0.5 mg monthly [34]. The results from these important studies demonstrated that all aflibercept regimens (and particularly injections every 2 months) were equivalent and demonstrated the noninferiority of aflibercept to ranibizumab in the treatment of nAMD, with fewer injections, reducing treatment burden for patients and caregivers [46].
ALTAIR was another RCT that compared the efficacy and safety of aflibercept in a T&E regimen with two different intervals of adjustment (2 or 4 weeks) [47]. It found improved functional and anatomic outcomes at week 52, maintained to week 96, with similar results in both treatment groups [47]. In this trial, 60% of patients reached a treatment interval of 12 weeks, and 40% of patients reached a treatment interval of 16 weeks, maintaining efficacy at 12 months (Table 3).
2.1.5 Brolucizumab
2.1.5.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
Brolucizumab (formerly ESBA1008 and RHT258, now Beovu®; Novartis, Basel, Switzerland) is a single-chain antibody fragment that inhibits the VEGF-A isoform. Its molecular structure is smaller (26 KDa) than other anti-VEGF agents, so brolucizumab can be much more concentrated [18, 19]. The serum half-life of brolucizumab is 5 days [18, 19]. Brolucizumab is highly stable and soluble, which allows administration of high doses in a single 50 μL intravitreal injection.
At a dose of 6 mg, its equivalent molar dose is approximately ten times greater than that of aflibercept and approximately 20 times greater than that of bevacizumab and ranibizumab [18, 19]. Preclinical data demonstrated an approximate twofold higher exposure in the retina and RPE/choroid, respectively, with brolucizumab, which may explain its better effect in reducing fluid accumulation in all retinal layers [48] (Table 2).
2.1.5.2 Efficacy and Registration Studies
A phase II trial, OSPREY, compared brolucizumab 6 mg and aflibercept 2 mg, recording anatomic advantages with brolucizumab and reaching noninferiority in BCVA [35].
Two phase III trials, HAWK and HARRIER, evaluated brolucizumab in two different doses (3 and 6 mg) compared with an aflibercept 2 mg fixed dosage in nAMD. These studies found noninferior outcomes in terms of BCVA and better anatomic outcomes with brolucizumab at week 48 [35]: a more stable central subfield thickness, with fewer unscheduled treatments and better fluid resolution than with aflibercept [19]. HAWK and HARRIER demonstrated that more than half of patients treated with brolucizumab maintained a 12-week regimen in the first year, confirming a longer half-life than aflibercept [35] (Table 3).
2.1.5.3 Local and Systemic Safety of Marketed Anti-VEGF Drugs
The systemic use of anti-VEGF drugs for the treatment of several cancers has been strongly associated with an increased risk of serious adverse reactions. Whether intravitreal administration of anti-VEGF drugs also carries this risk remains uncertain.
Studies showed that the frequency of severe systemic adverse events (SSAEs), including arterial thromboembolic events and death, was low with intravitreal administration [49]. A recent study explored the risk of systemic adverse events (AEs) after intravitreal bevacizumab, ranibizumab, and aflibercept in routine clinical practice and reported no differences in acute myocardial infarction, cardiovascular diseases, major bleedings, or hospitalization for all causes between the drugs [50].
More recently, using the Italian spontaneous reporting system [51], it was possible to provide an overview of the safety of anti-VEGF drugs in eye care settings. Specifically, of 2472 anti-VEGF drug-related reports, 299 (12.1%) were attributed to intravitreal use of these drugs. Most serious adverse drug reactions related to anti-VEGF drugs in patients with cancer are known and clinically relevant (e.g., gastrointestinal and vascular disorders). The frequency of reported serious adverse drug reactions was higher for intravitreal than for systemic use of anti-VEGF drugs in patients with cancer (58.9 vs. 34.1%; p < 0.001) and were disproportionally associated with ischemic heart disease and thromboembolic and cerebrovascular events.
In addition, VEGF regulates several features of the central nervous system, particularly in dopaminergic neurons, so that its inhibitors seem to be linked to Parkinson-like events and dementia. Two recent case reports described a potential link between intravitreal anti-VEGF use and Parkinson’s disease and dementia [52,53,54]. In a more recent study using VigiBase, a potential signal of an AE emerged as analyzing disproportionality by the preferred term “Parkinson’s disease” (N = 6 individual case safety reports; proportional reporting ratio 3.05 [95% confidence interval 1.36–6.81]) after intravitreal use of ranibizumab, which requires further investigation [52,53,54].
The adverse ocular effects with ranibizumab included endophthalmitis (from 0.5 to 0.9% at year 1 and 2, respectively), uveitis or other ocular inflammation, retinal and vitreous hemorrhages, elevated intraocular pressure (IOP) > 30 mmHg (ranging from 7.6% in the first year to 17.8% in the second year of treatment), and cataract compared with controls [55]. Similarly, the rates of serious ocular AEs at 1 and 2 years with bevacizumab were similar to those with ranibizumab for endophthalmitis, retinal detachment, traumatic cataract, and RPE tears, with bevacizumab having a higher rate of severe uveitis [31]. As reported in the post hoc analysis of CATT, ranibizumab and bevacizumab were both associated with an increased risk of developing geographic atrophy at 2-year FU (21 vs. 17%, respectively), specifically in case of monthly treatments versus a PRN regimen (24 vs. 15%, respectively) [39]. The incidence of ocular AEs with aflibercept was comparable to that with the other anti-VEGF drugs or were related to the injection procedure [55]. Data from pivotal studies reported brolucizumab was well-tolerated, with overall safety similar to that of aflibercept [35]. In the brolucizumab safety profile analysis, interesting AEs included uveitis and iritis (mild and moderate forms treated with topical corticosteroids; most resolved without any sequelae) [35, 56] and more severe cases of occlusive retinal vasculitis (RV) [55]. Specifically, intraocular inflammation was identified in 50 (4.6%) of the brolucizumab-treated patients in pivotal studies, among whom 36 (3.3%) had concomitant RV. The American Society of Retina Specialists conducted a post-approval analysis of brolucizumab-associated intraocular inflammation cases and concluded that, despite the risk of vision loss associated with RV following brolucizumab injection, the overall rate of vision loss in the study population was not different between the brolucizumab and aflibercept arms in HAWK and HARRIER [57]. On 27 May 2021, a notification of urgent safety measures in response to the increased incidence of intraocular inflammation (IOI) and related AEs, including RV and retinal vascular occlusion, in patients receiving doses every 4 weeks beyond the first three doses (“loading phase”) in nAMD was released, and studies with these regimens were terminated early (data on file; MERLIN first interpretable results. Novartis; 2021).
2.2 Non-EU-Approved Anti-VEGF Drugs
2.2.1 Conbercept
2.2.1.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
Conbercept (KH902) (Lumitin; Chengdu Kanghong Biotech Co, Ltd, Chengdu, China) is a 141-kDa engineered fusion protein produced by the gene recombination of VEGF receptor domain with Fc fragment of human Ig [58]. It is approved only in China in 2013 to treat nAMD [58]. Conbercept blocks all isoforms of VEGF-A, VEGF-B, VEGF-C, and PlGF [19, 59].
Animal studies reported a serum t½ of 4–5 days in monkey models [60]. During animal experiments, conbercept had a longer t½ and greater bioavailability than ranibizumab. In fact, the most relevant focus was to test the efficacy of conbercept in patients with nAMD using a less frequent maintenance dosing interval regimen [58].
2.2.1.2 Efficacy and Registration Studies
The phase II AURORA study assessed the safety and efficacy of conbercept 0.5 versus 2.0 mg monthly or PRN in patients with nAMD and reported that the treatment was well-tolerated at month 12 [61].
The Chinese registration phase III study (PHOENIX) was a 12-month prospective RCT that enrolled patients with nAMD, including polypoidal choroidal vasculopathy without subfoveal atrophy or scarring. The treatment protocol regimen was as follows: the conbercept group received a loading dose of conbercept 0.5 mg every month for 3 months then an injection every 3 months until month 12; the sham group received no injections in the first 3 months, then conbercept was administered monthly for 3 months, followed by an injection every 3 months until month 12 [58].
At month 3 in the conbercept group, 49.4% of patients gained ≥ 10 letters and 23.5% gained ≥ 15 letters in BCVA, with a significant improvement in anatomic outcomes compared with sham; at month 12, no statistically significant differences were reported for functional or anatomic outcomes.
Two other phase III RCTs are ongoing: PANDA-1 and PANDA-2 (randomized quadruple-blinded multicenter studies) are comparing conbercept 0.5 or 1.0 mg versus aflibercept 2.0 mg, respectively. In these studies, after the first two monthly doses, patients with nAMD receive doses every 2 months for 36 weeks [62]. In April 2021, the week-36 primary endpoint evaluation for the PANDA global clinical studies was available, and the desired primary endpoint was not met in either PANDA study. The analysis showed that no safety signals were detected. After consultation with the steering committee, Kanghong discontinued the PANDA studies.
2.2.1.3 Safety
Conbercept was well-tolerated intravitreally, and the PHOENIX study reported similar rates of ocular and systemic AEs for both treated and sham groups, and no serious AEs (SAEs) were reported. The most common ocular AEs were increase in IOP and conjunctival hemorrhage. The large molecular size of conbercept limits the permeability through blood–ocular barriers, reducing systemic exposure [58]. Interestingly, conbercept seemed to reduce the plasma level of VEGF more effectively than did ranibizumab [59].
A recent systematic review reported that conbercept produced a change in BCVA similar to that of ranibizumab at 3-month FU, with a better reduction in central retinal thickness (CRT), probably due to the different molecular target of these two molecules [59].
2.2.2 Abicipar
2.2.2.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
Abicipar pegol (AGN-150998, MP0112; Allergan plc, Dublin, Ireland/Molecular Partners, Switzerland) is an anti-VEGF molecule based on the designed ankyrin repeat proteins (DARPin) family, binding VEGF-A [62]. DARPin molecules are designed to be smaller with high selectivity and to be active at lower concentrations with increased stability, affinity, and specificity [55, 56].
The specific molecular characteristics include the low Mw (only 34 kDa, consisting of a recombinant DARPin protein of 14 kDa plus a 20 kDa polyethylene glycol portion) and the high target-binding affinity (Kd 1–4 pmol/L) [55, 63]. The t½ in aqueous humor was 13 days, longer than that of ranibizumab [19].
2.2.2.2 Efficacy and Registration Studies
Phase II and III studies compared abicipar with ranibizumab, in particular in the parallel, randomized, double-masked, multicenter CEDAR and SEQUOIA trials, which compared two different dosing interval regimens of abicipar 2.0 mg (every 8 weeks [Q8W] and every 12 weeks [Q12W]) versus ranibizumab 0.5 mg monthly (Q4W) through week 96, in treatment-naïve patients with nAMD. Abicipar maintained stable vision in > 91% of patients receiving the Q12W regimen [62], and BCVA improvement was sustained during the second year [64, 65]. The mean CRT was significantly reduced in all treatment groups in the first year, and only four intravitreal injections of abicipar were required to maintain anatomical benefit in the second year [64, 65].
2.2.2.3 Safety
CEDAR and SEQUOIA revealed a higher IOI, such as uveitis, vitreitis, and RV, and a higher incidence of endophthalmitis in the abicipar group compared with ranibizumab for both treatment doses. In detail, intraocular inflammation in the study eye was reported for 96 patients (15.4%) in the abicipar Q8W group, 96 patients (15.3%) in the abicipar Q12W group, and two patients (0.3%) in the ranibizumab Q4W group. As this effect may have been due to molecular impurities in the manufacturing of DARPin, another reformulated agent was used in the phase II MAPLE trial to test the safety of this molecule [62]. The improved manufacturing process led to an IOI of 8.9%, which was lower than that observed in the previous phase III studies.
Considering all these data, the US FDA considered the benefit:risk ratio unfavorable in terms of the rate of ocular inflammation with the use of abicipar pegol 2 mg/0.05 mL in patients with nAMD [55].
2.2.3 Pegpleranib
2.2.3.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
Pegpleranib (Fovista; Ophthotech Corp., New York, NY, USA) is a platelet-derived growth factor (PDGF) inhibitor that blocks the interaction between PDGF and its receptor. It was administered with other anti-VEGF agents (ranibizumab, bevacizumab, or aflibercept), and this combined mechanism could act on the vascular wall and particularly on pericytes, allowing a better response to anti-VEGF [66].
2.2.3.2 Efficacy and Registration Studies
Despite encouraging results from phase II studies, which reported a 62% benefit versus anti-VEGF, data from the phase III trial showed no statistically significant differences in BCVA between monotherapy or combined groups [19]. The addition of pegpleranib 1.5 mg to an aflibercept or bevacizumab regimen did not result in benefit as measured by the mean change in BCVA at the 12-month time point.
2.3 Agents under Investigation
2.3.1 Faricimab
2.3.1.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
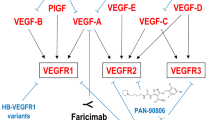
Faricimab (formerly RG7716; Roche/Genentech) is a bi-specific antibody that blocks VEGF-A and angiopoietin-2 (Ang-2). Levels of Ang-2 are elevated during nAMD, so simultaneous blockage of Ang-2 may lead to magnification of the anti-inflammatory effect [62]. Its molecular structure contains a modified Fc portion protecting against systemic absorption and intraocular inflammation [56].
2.3.1.2 Efficacy and Registration Studies
STAIRWAY was a phase II noninferiority study evaluating faricimab 6 mg (Q4W × 4 doses, then Q12W and Q16W) compared with ranibizumab 0.5 mg Q4W. It reported similar BCVA results between groups at week 52 [56, 62]. The protocol provided that patients in the Q16W regimen at week 24 could be switched to Q12W dosing according to predefined activity criteria; the analysis of results reported that, at week 24, 65% of patients had no disease activity [55], suggesting a sustained long interval dose regimen effect for the majority of patients.
Another phase II randomized double-masked multicenter study, AVENUE, enrolled treatment-naïve patients with nAMD and evaluated ranibizumab 0.5 mg Q4W and Q4W × 3 followed by faricimab 6 mg Q4W, respectively, compared with faricimab in different doses and interval regimens: faricimab 1.5 mg Q4W or 6 mg Q4W and Q4W × 4 followed by Q8W. In all arms, a significant reduction of CRT was reported, which was more relevant in the combination arm, leading to conclusions about the efficacy and safety of faricimab Q4W and Q8W compared with ranibizumab Q4W [62].
Two phase III triple-masked parallel RCTs, TENAYA and LUCERNE, are currently exploring faricimab Q16W versus aflibercept Q8W [56, 62]. On 25 January 2021, Roche announced that both studies had met their primary endpoints and showed that people receiving faricimab injections at fixed intervals of up to Q16W achieved visual acuity outcomes that were noninferior to those receiving aflibercept injections Q8W. Nearly half (45%) of people in both studies were treated with faricimab Q16W during the first year with a good safety profile. The nAMD open-label extension study is ongoing.
2.3.1.3 Safety
Phase II studies reported no significant differences in terms of AEs between faricimab and other anti-VEGF agents [56] (Table 4).
2.3.2 Port Delivery System
2.3.2.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
The port delivery system (PDS) (Genentech) is a novel permanent refillable drug reservoir that releases ranibizumab from a device surgically placed in the pars plana [19, 56, 62].
The mechanism of action of this device is a sustained release of anti-VEGF agent for passive diffusion from the implant into the vitreous [62]. This kind of device may reduce patient treatment burden by avoiding monthly injections.
2.3.2.2 Efficacy and Registration Studies
The phase II LADDER study was an RCT that tested the efficacy and safety of this device in patients with nAMD, evaluating ranibizumab in PDS (10, 40, and 100 mg/mL) compared with monthly ranibizumab 0.5 mg intravitreal injections, respectively. The results from the PDS 100 mg/mL arm seemed to be comparable to those from the intravitreal ranibizumab arm [67, 68].
The phase III Archway study evaluated fixed-interval dosing: the PDS was refilled every 24 weeks with ranibizumab 100 mg/mL and compared with monthly injections of ranibizumab 10 mg/mL. Results demonstrated that 98.4% of patients using the PDS were able to go 6 months without needing additional treatment and achieved vision outcomes equivalent to patients receiving monthly ranibizumab eye injections, a current standard of care. In the study, PDS was generally well-tolerated, with a favorable benefit–risk profile. A phase IIIb study (MR42410) on the effectiveness and safety of a 36-week refill regimen for the PDS with ranibizumab versus aflibercept T&E in nAMD Q16W is ongoing.
2.3.2.3 Safety
The rate of vitreous hemorrhage was high during the surgical procedure (almost 50%). A novel surgical approach was tested to reduce this ocular AE, involving a scleral dissection to the pars plana followed by cauterization of the choroid, reducing the rate of postoperative vitreous hemorrhage to 4.5% [62]. No long-term studies were available to evaluate the local effects and durability of the implant (Table 4).
2.3.3 OPT-302
2.3.3.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
OPT-302 (OPTHEA Ltd.; Melbourne, Australia) is a VEGF-C/D inhibitor molecule. Reports from animal studies indicated beneficial effects in conjunction with other anti-VEGF drugs (binding VEGF-A) for the concomitant blockage of different isoforms of VEGF [62].
2.3.3.2 Efficacy and Registration Studies
Data from phase I/II studies reported the noninferiority of OPT-30 as monotherapy or in combination with other anti-VEGF drugs [69].
In the phase II study (NCT03345082), patients with nAMD were randomized to receive OPT-302 2 mg + ranibizumab 0.5 mg or OPT-302 0.5 mg + ranibizumab 0.5 mg or ranibizumab 0.5 mg alone. Phase II studies showed that the combination of OPT-302 + ranibizumab determined a significant improvement in BCVA, retinal fluid reduction, and lesion area at week 24 compared with ranibizumab monotherapy [62].
Two different phase III RCTs are currently evaluating the efficacy and safety of intravitreal OPT-302 in combination with aflibercept (COAST) or ranibizumab (ShOre) in patients with nAMD.
2.3.3.3 Safety
Safety data reported no differences between OPT-302 + ranibizumab compared with ranibizumab monotherapy (Table 4).
2.3.4 KSI-301
2.3.4.1 Molecular Characteristics, Pharmacodynamics, and Pharmacokinetics
KSI-301 (Kodiak Sciences; Palo Alto, CA, USA) is an anti-VEGF antibody polymer conjugate of humanized anti-VEGF monoclonal antibody; the presence of a phosphorylcholine-based polymer increases the stability. The molecular characteristics consist of ameliorated pharmacokinetics, with greater long-term concentration and bioactivity than standard of care [62].
2.3.4.2 Efficacy and Registration Studies
DAZZLE is an ongoing phase IIb/III randomized, double-masked, prospective study evaluating the efficacy and the safety of KSI-301 in treatment-naïve patients with nAMD compared with standard of care. Patients will receive KSI-301 5 mg in an individualized regimen every 12, 16, and 20 weeks after a monthly loading dose versus aflibercept 2 mg Q8W after three monthly loading doses [70].
2.3.4.3 Safety
No SAEs were reported, and phase I studies reported primary safety and good tolerability [62] (Table 4).
2.3.5 ADVM-022
ADVM-022 (AAV.7m8-aflibercept) is a recombinant, replication-deficient adeno-associated virus (AAV.7m8) gene therapy vector carrying a coding sequence for aflibercept. The phase I OPTIC trial and its long-term extension are currently active in patients with nAMD. Preliminary results showed that the patients in cohort 1 (6 × 1011 vg/eye) did not need any supplemental injections for ≥ 15 months. Patients in the low-dose cohorts 2 and 3 (2 × 1011 vg/eye) required a few rescue injections, and the safety profile was acceptable [71, 72].
2.3.6 RGX-314
RGX-314 (Regen BioPharma; La Mesa, CA, USA) uses a novel AAV8 vector to deliver a genome that induces the production of an anti-VEGF Fab, similar to ranibizumab, delivered with vitrectomy and subretinal injection.
The phase II randomized dose-finding trial (AAVIATE) aims to evaluate the efficacy and safety of RGX-314 gene therapy in subjects with nAMD. Approximately 40 participants were planned to be enrolled into two cohorts with different doses in comparison with ranibizumab.
2.3.7 GB-102
GB-102 (GrayBug Vision; Redwood City, CA, USA) is an injectable form of sunitinib maleate, a tyrosine kinase inhibitor (TKI) that targets VEGF-A, PDGF, and many other kinases [73]. When injected, it forms a depot in the inferior vitreous that gradually biodegrades over time. Available data supported the finding that GB-102 treatment can last up to 6 months with stable visual acuity and retinal thickness outcomes before another dose is necessary.
ADAGIO (NCT03249740) was a phase I/IIa open-label single-dose trial of GB-102 in patients with nAMD that met the primary endpoints of safety and tolerability.
ALTISSIMO is an ongoing phase IIb (NCT03953079) randomized single-masked controlled study evaluating the safety and duration of effect of GB-102 as measured by time to first rescue treatment across several dose levels of GB-102 administered every 6 months as compared with intravitreal aflibercept administered every 2 months in subjects with nAMD who have received prior induction with anti-VEGF. According to the preliminary results, nAMD eyes treated with the 2-mg dose were all switched to the 1-mg dose of GB-102.
2.3.8 EYP-1901
EYP-1901 is a potential twice-yearly sustained-delivery intravitreal anti-VEGF treatment for nAMD. EYP-1901 combines a bioerodible formulation of EyePoint’s proprietary Durasert® sustained-release technology with vorolanib, a TKI. Vorolanib provided clear efficacy signals in two prior human trials in nAMD as an orally delivered therapy with no significant ocular AEs. The phase I DAVIO open-label dose-escalation trial will examine 13 patients who were responsive to previous anti-VEGF treatments with a single intravitreal injection of EYP-1901 [74].
2.3.9 CLS-AX
Axitinib is a TKI currently approved to treat renal cell cancer that achieves pan-VEGF blockade, directly inhibiting VEGF receptors 1, 2, and 3 with high potency and specificity. CLS-AX (axitinib injectable suspension) was administered by suprachoroidal injection via Clearside’s SCS Microinjector in six patients with wet AMD in a phase I/II open-label dose-escalation OASIS trial. The trial is ongoing [75].
2.3.10 PAN-90806
PAN-90806 (PanOptica; Mount Arlington, NJ, USA) is a TKI of VEGF-A and PDGF. This drug is delivered topically as eye drops that use the transscleral vascular route to reach the target tissues in the retina.
A phase I study confirmed the efficacy of the topical treatment. However, punctate keratopathy was a common adverse effect. The formulation was modified, and a subsequent phase I/II randomized, double-masked, uncontrolled study in treatment-naïve patients with nAMD using PAN-90806 topical drops found “no major or serious untoward (unfavorable and unintended) safety issues or trends” [76].
2.3.11 ICON-1
ICON-1 (Iconic Therapeutics; South San Francisco, CA, USA) is a recombinant modified factor VIIIa protein linked with the Fc portion of a human IgG1. It binds to tissue factor, which is overexpressed in CNV, but does not interfere with normal blood coagulation. A phase I open-label dose-escalation nonrandomized study of intravitreal injection of ICON-1 was performed in patients with nAMD (n = 18) [77, 78]. The primary endpoint of safety and tolerability was met, as no patients had any drug-related SAEs.
EMERGE was a phase II randomized double-masked study of intravitreal injections in patients with nAMD (n = 88) [77, 78], who were randomized to a combination of ranibizumab 0.5 mg and ICON-1 0.3 mg, ranibizumab 0.5 mg only, or ICON-1 0.3 mg only for 3 months. CNV decreased by 40% in the combination arm, 14.6% in the ranibizumab-only arm, and 17.2% in the ICON-1-only arm at 6 months, with the same improvement in BCVA and reduction of CRT in all groups. DECO (NCT03452527) was a phase II randomized open-label parallel study in patients with nAMD (n = 15). All patients received initial treatment with aflibercept and then maintenance therapy with intravitreal injection of ICON-1 0.6 mg or intravitreal injection of aflibercept 2 mg.
2.3.12 AKST4290
AKST4290 (formerly ALK4290; Alkahest; San Carlos, CA, USA) is an oral treatment that targets eotaxin, an immunomodulatory chemokine that is highly expressed in choroidal endothelial cells and in the circulation in patients with nAMD. The agent is an inhibitor against the natural receptor for eotaxin (CCR3). AKST4290–201 (NCT03558061) and AKST4290–202 (NCT03558074) were both phase IIa single-arm open-label studies of oral AKST2490 400 mg (twice daily) monotherapy in treatment-naïve patients with nAMD who no longer responded to anti-VEGF. AKST4290–201 and AKST4290–202 showed that 83 and 72% of patients, respectively, had stable or better BCVA at 6 weeks, with good safety. The PHTHALO-205 study was planned to evaluate the efficacy and safety of AKST4290 in combination with aflibercept injections in treatment-naïve subjects with nAMD.
2.3.13 DE-122
DE-122 (Carotuximab; Santen; Osaka, Japan/TRACON Pharmaceuticals; San Diego, CA, USA) is an antibody that binds endoglin, a protein with a critical role in angiogenesis. This drug is an ophthalmic reformulation of TRC 105, an anticancer drug made by TRACON.
PAVE (NCT02555306) was a phase I/II open-label dose-escalating sequential-cohort study of intravitreal DE-122 in patients with nAMD (n = 12) who were refractory to VEGF inhibitors. The study explored different doses and found an approximate two- or three-letter improvement and a 0.36–0.116 micron thickness reduction in the new drug arms. No medication-related SAEs had been observed at 90 days.
AVANTE (NCT03211234) is an ongoing phase II multicenter randomized double-masked active-control study of intravitreal injections of DE-122 in patients with nAMD (n = 76). Patients were randomized 1:1:1 to arms that received either DE-122 with ranibizumab, high-dose DE-122 with ranibizumab, or ranibizumab alone.
3 Biosimilar Anti-VEGF Drugs
A biosimilar is a copy of a biological agent defined as “a medical product with a similar safety, efficacy and quality as an already authorized biologic product” [79]. Since 2006 in Europe, biosimilar drugs have been marketed to improve access to care by offering biological drugs that are not clinically different from the originator, at a lower price, which is even more beneficial in developing countries [79, 80].
The overall cost and time for development and final marketing approval for a biosimilar is much less than for the originator; nevertheless, biosimilars undergo extensive premarketing comparability exercises with their originator, as required by regulatory authorities [81].
Recently, biosimilars of anti-VEGF agents were introduced for the treatment of retinal vascular diseases.
3.1 Ranibizumab Biosimilar Drugs
To date, there are seven biosimilars of ranibizumab.
Razumab® (Intas Pharmaceuticals Ltd; Ahmedabad, GJ, India) was the world’s first biosimilar to ranibizumab and was approved in 2015 by the Drug Controller General of India for the treatment of nAMD [80, 82]. A head-to-head study compared the efficacy and safety of razumab with that of innovator ranibizumab and showed similar relative binding and potency with some differences in the serine and asparagine sequences [83].
Two important retrospective multicenter observational studies, RE-ENACT and RE-ENACT 2 (a long-term extension of RE-ENACT), tested the use of this molecule in the treatment of retinal vascular disorders in a real-world setting, making conclusions about the safety and efficacy of this drug [80,81,82]. The RE-ENACT study enrolled 561 eyes affected by AMD, diabetic macular edema, and retinal vein occlusion for a FU period of 12 weeks; in the RE-ENACT 2 study, 341 eyes were enrolled with 48 weeks of FU, including eyes affected by myopic CNV [80, 82].
Data from the CESAR study showed a rapid BCVA improvement and reduction in retinal thickness, with efficacy observed as early as 1 month and maintained until 3 months [84].
These studies reported encouraging results in terms of improvement in BCVA accompanied by a stabilization or reduction of retinal thickness in patients with nAMD treated with razumab at weeks 12 and 48; the subgroup analysis found no differences related to lesion type [80].
The retrospective and real-life nature of these studies represented the major limitations. The ocular AEs were generally similar to those with ranibizumab, but a higher incidence of sterile intraocular inflammation was reported, probably due to a higher endotoxin level; for this reason, the molecule was revised by the manufacturer [80].
The prospective, multicenter ASSET study enrolled 126 patients with nAMD to receive intravitreal razumab 0.5 mg monthly for 24 weeks and tested the immunogenicity of this molecule; no AEs suggestive for immunogenicity were noted during the 6-month FU period [85].
Data from a major clinical real-world study were recently published. This study analyzed 6404 eyes treated with intravitreal injections of Razumab over 4.25 years for several retinal vascular diseases, including nAMD (15.29%), and with different treatment regimens (PRN or T&E) [86]. This important long-term FU study reported that the most common ocular AEs (1978 events/9406 injections performed during the study period) were subconjunctival hemorrhage (8.2%), transient blurring of vision (6.5%), and mild ocular pain (5.27%) and rare cases of raised IOP (0.33%), RPE tear (0.33%), mild anterior uveitis (0.1%), vitreitis (0.02%), and endophthalmitis (0.01%), with an incidence similar to that with ranibizumab. The incidence of nonfatal myocardial infarction and nonfatal cerebrovascular accident was 0.12 and 0.09%, respectively [86].
The following six biosimilars to ranibizumab have reached phase III studies.
-
R TPR 024 (Reliance Life Sciences Pvt Ltd; Navi Mumbai, MH, India) has completed the prospective multicenter double-masked phase III RCT, enrolling 159 patients with nAMD. This study compared R TPR 024 or innovative ranibizumab injected monthly for 24 weeks [81].
-
Lupin Ltd (Mumbai, India) started a phase III trial of ranibizumab that is ongoing to assess BCVA changes at week 8 during a treatment regimen of monthly injections for 3 months [81].
-
FYB201 (Formycon AG/Bioeq; Germany) has completed the COLUMBUS-AMD study, a quadruple-masked multicenter phase III trial comparing BCVA changes between groups with a FU period of 8 weeks. Similar efficacy between FYB201 and innovator ranibizumab was reported [81]. The data were resubmitted to the FDA for approval.
-
Xlucane (Xbrane Biopharma; Solna, Sweden) is being evaluated in the ongoing XPLORE phase III trial, a multicenter double-masked trial involving 580 nAMD eyes and analyzing changes in BCVA at week 8 between Xlucane or innovator ranibizumab administered monthly until 52 weeks [81].
-
SB11 (Byooviz; Samsung Bioepis Co Ltd; Incheon, South Korea) is being tested in a quadruple-masked multicenter phase III trial evaluating the pharmaceutical profile, immunogenicity, efficacy, and safety versus that of innovator ranibizumab in 705 patients with nAMD with a FU period of 52 weeks [81, 87]. Patients received SB11 0.5 mg or ranibizumab 0.5 mg intravitreally every 4 weeks for 48 weeks; the primary endpoints were BCVA changes from baseline to week 8 and anatomical features at week 4. Data to week 24 were recently published, and SB11 demonstrated equivalence for both primary endpoints with safety and immunogenicity profiles similar to those of reference ranibizumab [87]. Ad interim data on a small sample showed a rate of endophthalmitis similar to that with ranibizumab [86]. On 23 August 2021, the European Commission approved Byooviz, which will be sold in the EU by Biogen [88].
-
SJP-0133 (Senju Pharmaceuticals; Osaka, Japan) will complete the single-masked phase III RCT in 2022. In this study, patients are receiving three monthly injections of SJP-0133 or innovator ranibizumab followed by a particular PRN regimen from week 12 to week 48 instead of a fixed regimen, unlike the other drugs [81].
3.2 Bevacizumab Biosimilar Drugs
Bevacizumab biosimilars are already approved for clinical use in different countries after completion of phase III studies. In 2016, the Drug Controller General of India approved the following molecules: Cizumab (Hetero; Hyderabad, India), BevaciRel (Reliance Life Science; Mumbai, India), Zybev® (Zydus Cadilla; Ahmedabad, India), Bevatas® (Intas Pharmaceuticals; Ahmedabad, India), KRABEVA (Biocon; Bengaluru, India) [89]. The most used in ophthalmology were Zybev® and BevaciRel, which are less often used off label by retina specialists, as reported by the Vitreo-Retinal Society—India (VRSI), before the introduction of ranibizumab biosimilar drugs [90]. In Europe and the USA, ABP215 (MVASI; Allergan; Dublin Ireland/Amgen; Thousand Oaks, CA, USA) was approved in 2017 [89]. Two other bevacizumab biosimilar molecules were approved by the Russian regulatory body in 2015 and by the Argentina regulatory body in 2016: BCD-021 (Biocad; Saint Petersburg, Russia) and mAbxience (mAbxience; Madrid, Spain), respectively [89].
Recently, encouraging results with the use of Strivant® (CinnaGen Co., Iran), another bevacizumab biosimilar drug, in terms of BCVA and improved anatomical features were reported in a case series recruiting patients affected by several retinal disorders, including nAMD [91].
3.3 Aflibercept Biosimilar Drugs
To date, three biosimilars to aflibercept are being evaluated in phase III clinical trials for nAMD.
In Europe, FYB203 (Formycon AG; Munich, Germany/Bioeq GmbH; Holzkirchen, Germany) is being compared with innovator aflibercept in the MAGELLAN-AMD phase III study enrolling 400 patients with nAMD [81]. The study started in August 2020 with the objective of providing data for the efficacy, safety, and immunogenicity of FYB203 [92].
In the USA, ABP-938 (Amgen) is being evaluated for nAMD in a multicenter RCT comparing ABP-938 or innovator aflibercept bimonthly followed by a re-randomization for the aflibercept group at week 16, to switch in ABP-938 group until week 48, with a FU of 52 weeks [81].
In South Korea, SB15 (Samsung Bioepis Co, Ltd) is currently being compared with innovator aflibercept in an RCT enrolling 446 patients with nAMD. Subjects receive three monthly injections followed by a bimonthly regimen until week 48; at week 32, subjects in the aflibercept group will be re-randomized to receive SB15 or innovator aflibercept. The primary outcome is BCVA change from baseline at week 8 [81].
4 Public and Global Health Issues
With expanding indications of antiangiogenic drugs for AMD, as well as with the increasing number of drugs becoming available, drug expenditure in this therapeutic field has grown [93].
In recent years, much of the debate on antiangiogenic drugs for retinal vascular diseases has focused on the use of off-label bevacizumab instead of ranibizumab [94]. The debate on the efficacy and safety of bevacizumab versus ranibizumab for AMD started after CATT (an RCT sponsored by the National Institutes of Health comparing ranibizumab and bevacizumab published in 2011) found a similar efficacy of the two drugs but more SSAEs with bevacizumab [94]. At the time, bevacizumab (off-label) cost $US40 and ranibizumab (approved) cost $US2000. After a few years of discussion and polemics, based both on the sparse available data and on regulatory issues, a Cochrane systematic review [95] of non-industry-sponsored RCTs did not detect a difference between intravitreal bevacizumab and ranibizumab for deaths, all SSAEs, or specific subsets of SSAEs in the first 2 years of treatment, with the exception of gastrointestinal disorders.
Safety related to the sterility of the production chain of off-label bevacizumab was also of great concern in India after counterfeit bevacizumab had caused outbreaks of sterile and infectious post-injection endophthalmitis in several countries, leading to a temporary halt of its authorization for ophthalmic use [96]. A recent study found that in-house compounded injections of bevacizumab can reduce post-injection endophthalmitis to a minimum with maintenance of proper asepsis and strict protocols by the compounding pharmacy [97].
As noted in Sect. 3, biosimilar drugs have been developed to contain drug costs for the treatment of a range of diseases, including retinal vascular conditions. The fact that many of these drugs are produced locally and there is greater flexibility for drug approval makes India and some East Asian countries open to earlier testing of new biosimilar drugs than the USA and Europe [81]. Cost is an important determinant of medical decision making and drug selection in most countries, and this makes a shift towards less expensive biosimilars an attractive strategy. The VRSI survey established that Indian physicians are well aware of biosimilars and that there is an increasing trend toward prescribing a ranibizumab biosimilar [90]. A third of the respondents felt that Razumab is appropriately priced, but they acknowledged that a further price reduction would be necessary for Razumab to become the drug of first choice. The US patents on Lucentis and Eylea will end in 2020, whereas the European patents expire in 2022 and 2025, respectively. With patents of these biologics about to expire, and the growing acceptance of biosimilars, a shift from branded drugs toward biosimilars in developed nations may occur.
Key issues regarding drug effectiveness with biologics for long-term conditions are the ability to deliver the intravitreal injections in a way that not only minimizes costs but also, and first of all, maximizes benefits. This means that logistics and human resource problems may have the same impact as an ineffective drug on health outcomes. In fact, both undertreatment and noncompliance are common in low- and middle-income countries and in industrialized countries [98, 99]. In age-related conditions, compliance is an issue since the burden of care from multimorbidity and polytherapy is substantial. Despite all these concerns, baseline vision can be maintained on average if patients are followed and injected regularly, as shown in a UK study, which also found that, at 10 years, one-third of eyes had a level of vision that allowed driving (20/40) [100].
Equity issues also affect the appropriateness of the delivery of intravitreal injections, even in industrialized countries. A study conducted in England found wide variation in the provision of anti-VEGF intravitreal injections across services [101]. The occurrence of AMD is in itself related to education, employment, and household income, independently of cardiovascular risk factors [102].
5 Conclusions
The effectiveness and safety of intravitreal drugs for AMD is related to not only drug properties but also factors that affect the provision of services in clinical pathways, and the interaction between patients and services is as critical as efficacy and safety when maximizing health outcomes for these patients.
References
Mitchell P, Liew G, Gopinath B, Wong TY. Age-related macular degeneration. Lancet. 2018;392(10153):1147–59.
Chew EY, Clemons TE, Agrón E, Sperduto RD, Sangiovanni JP, Davis MD, Ferris FL 3rd. Ten-year follow-up of age-related macular degeneration in the age-related eye disease study: AREDS report no. 36. JAMA Ophthalmol. 2014;132(3):272–7.
Gangnon RE, Lee KE, Klein BE, Iyengar SK, Sivakumaran TA, Klein R. Severity of age-related macular degeneration in 1 eye and the incidence and progression of age-related macular degeneration in the fellow eye: the Beaver Dam Eye Study. JAMA Ophthalmol. 2015;133(2):125–32.
Shin HT, Yoon BW, Seo JH. Comparison of risk allele frequencies of single nucleotide polymorphisms associated with age-related macular degeneration in different ethnic groups. BMC Ophthalmol. 2021;21(1):97.
Jones M, Whitton C, Tan AG, Holliday EG, Oldmeadow C, Flood VM, Sim X, Chai JF, Hamzah H, Klein R, et al. Exploring factors underlying ethnic difference in age-related macular degeneration prevalence. Ophthalmic Epidemiol. 2020;27(5):399–408.
Spaide RF, Jaffe GJ, Sarraf D, Freund KB, Sadda SR, Staurenghi G, Waheed NK, Chakravarthy U, Rosenfeld PJ, Holz FG, et al. Consensus nomenclature for reporting neovascular age-related macular degeneration data: consensus on neovascular age-related macular degeneration nomenclature study group. Ophthalmology. 2020;127(5):616–36.
Assi L, Chamseddine F, Ibrahim P, Sabbagh H, Rosman L, Congdon N, Evans J, Ramke J, Kuper H, Burton MJ, et al. A global assessment of eye health and quality of life: a systematic review of systematic reviews. JAMA Ophthalmol. 2021;139:526.
Li E, Donati S, Lindsley KB, Krzystolik MG, Virgili G. Treatment regimens for administration of anti-vascular endothelial growth factor agents for neovascular age-related macular degeneration. Cochrane Database Syst Rev. 2020;5:CD012208.
Mitchell SL, Uppal K, Williamson SM, Liu K, Burgess LG, Tran V, Umfress AC, Jarrell KL, Cooke Bailey JN, Agarwal A, et al. The carnitine shuttle pathway is altered in patients with neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci. 2018;59(12):4978–85.
Fogli S, Del Re M, Rofi E, Posarelli C, Figus M, Danesi R. Clinical pharmacology of intravitreal anti-VEGF drugs. Eye (Lond). 2018;32(6):1010–20.
D'Amico DJ, Masonson HN, Patel M, Adamis AP, Cunningham ET Jr, Guyer DR, Katz; VEGF Inhibition Study in Ocular Neovascularization (V.I.S.I.O.N.) Clinical Trial Group. Pegaptanib sodium for neovascular age-related macular degeneration: two-year safety results of the two prospective, multicenter, controlled clinical trials. Ophthalmology. 2006;113(6):992–1001.e6.
Bakri SJ, Snyder MR, Reid JM, Pulido JS, Ezzat MK, Singh RJ. Pharmacokinetics of intravitreal ranibizumab (Lucentis). Ophthalmology. 2007;114(12):2179–82.
Christoforidis JB, Briley K, Binzel K, Bhatia P, Wei L, Kumar K, Knopp MV. Systemic biodistribution and intravitreal pharmacokinetic properties of bevacizumab, ranibizumab, and aflibercept in a nonhuman primate model. Invest Ophthalmol Vis Sci. 2017;58(13):5636–45.
Gaudreault J, Fei D, Rusit J, Suboc P, Shiu V. Preclinical pharmacokinetics of Ranibizumab (rhuFabV2) after a single intravitreal administration. Invest Ophthalmol Vis Sci. 2005;46(2):726–33.
Zhu Q, Ziemssen F, Henke-Fahle S, Tatar O, Szurman P, Aisenbrey S, Schneiderhan-Marra N, Xu X, Tubingen Bevacizumab Study G, Grisanti S. Vitreous levels of bevacizumab and vascular endothelial growth factor-A in patients with choroidal neovascularization. Ophthalmology. 2008;115(10):1750–5 (1755 e1751).
Avery RL, Castellarin AA, Steinle NC, Dhoot DS, Pieramici DJ, See R, Couvillion S, Nasir MA, Rabena MD, Maia M, et al. Systemic pharmacokinetics and pharmacodynamics of intravitreal aflibercept, bevacizumab, and ranibizumab. Retina. 2017;37(10):1847–58.
Avery RL, Castellarin AA, Steinle NC, Dhoot DS, Pieramici DJ, See R, Couvillion S, Nasir MA, Rabena MD, Le K, et al. Systemic pharmacokinetics following intravitreal injections of ranibizumab, bevacizumab or aflibercept in patients with neovascular AMD. Br J Ophthalmol. 2014;98(12):1636–41.
Holz FG, Dugel PU, Weissgerber G, Hamilton R, Silva R, Bandello F, Larsen M, Weichselberger A, Wenzel A, Schmidt A, et al. Single-chain antibody fragment VEGF inhibitor RTH258 for neovascular age-related macular degeneration: a randomized controlled study. Ophthalmology. 2016;123(5):1080–9.
Kniggendorf V, Dreyfuss JL, Regatieri CV. Age-related macular degeneration: a review of current therapies and new treatments. Arq Bras Oftalmol. 2020;83(6):552–61.
Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY, Group MS. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–31.
Kaiser PK, Blodi BA, Shapiro H, Acharya NR, Group MS. Angiographic and optical coherence tomographic results of the MARINA study of ranibizumab in neovascular age-related macular degeneration. Ophthalmology. 2007;114(10):1868–75.
Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, Sy JP, Schneider S, Group AS. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1432–44.
Brown DM, Michels M, Kaiser PK, Heier JS, Sy JP, Ianchulev T, Group AS. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR study. Ophthalmology. 2009;116(1):57-65 e55.
Singer MA, Awh CC, Sadda S, Freeman WR, Antoszyk AN, Wong P, Tuomi L. HORIZON: an open-label extension trial of ranibizumab for choroidal neovascularization secondary to age-related macular degeneration. Ophthalmology. 2012;119(6):1175–83.
Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR, Zhang K. Group S-US: seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology. 2013;120(11):2292–9.
Regillo CD, Brown DM, Abraham P, Yue H, Ianchulev T, Schneider S, Shams N. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER Study year 1. Am J Ophthalmol. 2008;145(2):239–48.
Abraham P, Yue H, Wilson L. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER study year 2. Am J Ophthalmol. 2010;150(3):315-324 e311.
Ho AC, Busbee BG, Regillo CD, Wieland MR, Van Everen SA, Li Z, Rubio RG, Lai P, Group HS. Twenty-four-month efficacy and safety of 0.5 mg or 2.0 mg ranibizumab in patients with subfoveal neovascular age-related macular degeneration. Ophthalmology. 2014;121(11):2181–92.
Wykoff CC, Croft DE, Brown DM, Wang R, Payne JF, Clark L, Abdelfattah NS, Sadda SR, Group T-AS. Prospective trial of treat-and-extend versus monthly dosing for neovascular age-related macular degeneration: TREX-AMD 1-year results. Ophthalmology. 2015;122(12):2514–22.
Group CR, Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364(20):1897–908.
Comparison of Age-related Macular Degeneration Treatments Trials Research G, Martin DF, Maguire MG, Fine SL, Ying GS, Jaffe GJ, Grunwald JE, Toth C, Redford M, Ferris FL 3rd. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012;119(7):1388–98.
Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Culliford LA, Reeves BC. investigators is: alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet. 2013;382(9900):1258–67.
Tufail A, Patel PJ, Egan C, Hykin P, da Cruz L, Gregor Z, Dowler J, Majid MA, Bailey C, Mohamed Q, et al. Bevacizumab for neovascular age related macular degeneration (ABC Trial): multicentre randomised double masked study. BMJ. 2010;340:c2459.
Heier JS, Brown DM, Chong V, Korobelnik JF, Kaiser PK, Nguyen QD, Kirchhof B, Ho A, Ogura Y, Yancopoulos GD, et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology. 2012;119(12):2537–48.
Dugel PU, Koh A, Ogura Y, Jaffe GJ, Schmidt-Erfurth U, Brown DM, Gomes AV, Warburton J, Weichselberger A, Holz FG, et al. HAWK and HARRIER: phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology. 2020;127(1):72–84.
Yu L, Liang XH, Ferrara N. Comparing protein VEGF inhibitors: in vitro biological studies. Biochem Biophys Res Commun. 2011;408(2):276–81.
Bakri SJ, Snyder MR, Reid JM, Pulido JS, Singh RJ. Pharmacokinetics of intravitreal bevacizumab (Avastin). Ophthalmology. 2007;114(5):855–9.
Nomoto H, Shiraga F, Kuno N, Kimura E, Fujii S, Shinomiya K, Nugent AK, Hirooka K, Baba T. Pharmacokinetics of bevacizumab after topical, subconjunctival, and intravitreal administration in rabbits. Invest Ophthalmol Vis Sci. 2009;50(10):4807–13.
Comparison of Age-related Macular Degeneration Treatments Trials Research G, Maguire MG, Martin DF, Ying GS, Jaffe GJ, Daniel E, Grunwald JE, Toth CA, Ferris FL 3rd, Fine SL. Five-year outcomes with anti-vascular endothelial growth factor treatment of neovascular age-related macular degeneration: the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2016;123(8):1751–61.
Schauwvlieghe AM, Dijkman G, Hooymans JM, Verbraak FD, Hoyng CB, Dijkgraaf MG, Peto T, Vingerling JR, Schlingemann RO. Comparing the effectiveness of bevacizumab to ranibizumab in patients with exudative age-related macular degeneration. The BRAMD Study. PLoS ONE. 2016;11(5):e0153052.
Kodjikian L, Souied EH, Mimoun G, Mauget-Faysse M, Behar-Cohen F, Decullier E, Huot L, Aulagner G, Group GS. Ranibizumab versus bevacizumab for neovascular age-related macular degeneration: results from the GEFAL noninferiority randomized trial. Ophthalmology. 2013;120(11):2300–9.
Ehlers JP. The MANTA 1-year results: the anti-VEGF debate continues. Br J Ophthalmol. 2013;97(3):248–50.
Garcia-Quintanilla L, Luaces-Rodriguez A, Gil-Martinez M, Mondelo-Garcia C, Maronas O, Mangas-Sanjuan V, Gonzalez-Barcia M, Zarra-Ferro I, Aguiar P, Otero-Espinar FJ, et al. Pharmacokinetics of intravitreal anti-VEGF drugs in age-related macular degeneration. Pharmaceutics. 2019;11(8):365.
Kaiser PK, Kodjikian L, Korobelnik JF, Winkler J, Torri A, Zeitz O, Vitti R, Ahlers C, Zimmermann T, Dicioccio AT, et al. Systemic pharmacokinetic/pharmacodynamic analysis of intravitreal aflibercept injection in patients with retinal diseases. BMJ Open Ophthalmol. 2019;4(1):e000185.
Christoforidis JB, Williams MM, Kothandaraman S, Kumar K, Epitropoulos FJ, Knopp MV. Pharmacokinetic properties of intravitreal I-124-aflibercept in a rabbit model using PET/CT. Curr Eye Res. 2012;37(12):1171–4.
Schmidt-Erfurth U, Kaiser PK, Korobelnik JF, Brown DM, Chong V, Nguyen QD, Ho AC, Ogura Y, Simader C, Jaffe GJ, et al. Intravitreal aflibercept injection for neovascular age-related macular degeneration: ninety-six-week results of the VIEW studies. Ophthalmology. 2014;121(1):193–201.
Ohji M, Takahashi K, Okada AA, Kobayashi M, Matsuda Y, Terano Y, Investigators A. Efficacy and safety of intravitreal aflibercept treat-and-extend regimens in exudative age-related macular degeneration: 52- and 96-week findings from ALTAIR: a randomized controlled trial. Adv Ther. 2020;37(3):1173–87.
Holz FG, Tadayoni R, Beatty S, Berger AR, Cereda MG, Hykin P, Staurenghi G, Wittrup-Jensen K, Nilsson J, Kim K, et al. Determinants of visual acuity outcomes in eyes with neovascular AMD treated with anti-VEGF agents: an instrumental variable analysis of the AURA study. Eye (Lond). 2016;30(8):1063–71.
Solomon SD, Lindsley K, Vedula SS, Krzystolik MG, Hawkins BS. Anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Cochrane Database Syst Rev. 2019;3:CD005139.
Maloney MH, Payne SR, Herrin J, Sangaralingham LR, Shah ND, Barkmeier AJ. Risk of systemic adverse events after intravitreal bevacizumab, ranibizumab, and aflibercept in routine clinical practice. Ophthalmology. 2021;128(3):417–24.
Cutroneo PM, Giardina C, Ientile V, Potenza S, Sottosanti L, Ferrajolo C, Trombetta CJ, Trifiro G. Overview of the safety of anti-VEGF drugs: analysis of the Italian spontaneous reporting system. Drug Saf. 2017;40(11):1131–40.
Trifiro G, Marciano I, Cutroneo PM, Spina E, Mirabelli E, Trombetta CJ, Morgante F. Long-term intravitreal ranibizumab as a potential additional risk factor for neurodegeneration in Parkinson’s disease: a case report. Front Pharmacol. 2018;9:608.
Sultana J, Scondotto G, Cutroneo PM, Morgante F, Trifiro G. Intravitreal anti-VEGF drugs and signals of dementia and Parkinson-like events: analysis of the VigiBase database of spontaneous reports. Front Pharmacol. 2020;11:315.
Meyer PWUM, Schröder J. Intraocular injection of anti-VEGF agents. J Neurol Res Ther. 2018;2(3):10–3.
Ricci F, Bandello F, Navarra P, Staurenghi G, Stumpp M, Zarbin M. Neovascular age-related macular degeneration: therapeutic management and new-upcoming approaches. Int J Mol Sci. 2020;21(21):8242.
Ammar MJ, Hsu J, Chiang A, Ho AC, Regillo CD. Age-related macular degeneration therapy: a review. Curr Opin Ophthalmol. 2020;31(3):215–21.
ReST committee drug safety update. Presented at: American Society of Retina Specialists (ASRS) 2020 Virtual Annual Meeting; July 24–26, 2020.
Liu K, Song Y, Xu G, Ye J, Wu Z, Liu X, Dong X, Zhang M, Xing Y, Zhu S, et al. Conbercept for treatment of neovascular age-related macular degeneration: results of the randomized phase 3 PHOENIX study. Am J Ophthalmol. 2019;197:156–67.
Zhang J, Liang Y, Xie J, Li D, Hu Q, Li X, Zheng W, He R. Conbercept for patients with age-related macular degeneration: a systematic review. BMC Ophthalmol. 2018;18(1):142.
Zhang M, Yu D, Yang C, Xia Q, Li W, Liu B, Li H. The pharmacology study of a new recombinant human VEGF receptor-fc fusion protein on experimental choroidal neovascularization. Pharm Res. 2009;26(1):204–10.
Li X, Xu G, Wang Y, Xu X, Liu X, Tang S, Zhang F, Zhang J, Tang L, Wu Q, et al. Safety and efficacy of conbercept in neovascular age-related macular degeneration: results from a 12-month randomized phase 2 study: AURORA study. Ophthalmology. 2014;121(9):1740–7.
Samanta A, Aziz AA, Jhingan M, Singh SR, Khanani AM, Chhablani J. Emerging therapies in neovascular age-related macular degeneration in 2020. Asia Pac J Ophthalmol (Phila). 2020;9(3):250–9.
Rodrigues GA, Mason M, Christie LA, Hansen C, Hernandez LM, Burke J, Luhrs KA, Hohman TC. Functional characterization of abicipar-pegol, an anti-VEGF DARPin therapeutic that potently inhibits angiogenesis and vascular permeability. Invest Ophthalmol Vis Sci. 2018;59(15):5836–46.
Khurana RN, Kunimoto D, Yoon YH, Wykoff CC, Chang A, Maturi RK, Agostini H, Souied E, Chow DR, Lotery AJ, et al. Two-year results of the phase 3 randomized controlled study of abicipar in neovascular age-related macular degeneration. Ophthalmology. 2020;128:1027–38.
Kunimoto D, Yoon YH, Wykoff CC, Chang A, Khurana RN, Maturi RK, Agostini H, Souied E, Chow DR, Lotery AJ, et al. Efficacy and safety of abicipar in neovascular age-related macular degeneration: 52-week results of phase 3 randomized controlled study. Ophthalmology. 2020;127(10):1331–44.
Jaffe GJ, Eliott D, Wells JA, Prenner JL, Papp A, Patel S. A phase 1 study of intravitreous E10030 in combination with ranibizumab in neovascular age-related macular degeneration. Ophthalmology. 2016;123(1):78–85.
Campochiaro PA, Marcus DM, Awh CC, Regillo C, Adamis AP, Bantseev V, Chiang Y, Ehrlich JS, Erickson S, Hanley WD, et al. The port delivery system with ranibizumab for neovascular age-related macular degeneration: results from the randomized phase 2 ladder clinical trial. Ophthalmology. 2019;126(8):1141–54.
Khanani AM, Callanan D, Dreyer R, Chen S, Howard JG, Hopkins JJ, Lin CY, Lorenz-Candlin M, Makadia S, Patel S, et al. End-of-study results for the ladder phase 2 trial of the port delivery system with ranibizumab for neovascular age-related macular degeneration. Ophthalmol Retina. 2020;5:775–87.
Dugel PU, Boyer DS, Antoszyk AN, Steinle NC, Varenhorst MP, Pearlman JA, Gillies MC, Finger RP, Baldwin ME, Leitch IM. Phase 1 study of OPT-302 inhibition of vascular endothelial growth factors C and D for neovascular age-related macular degeneration. Ophthalmol Retina. 2020;4(3):250–63.
Chandrasekaran PR, Madanagopalan VG. KSI-301: antibody biopolymer conjugate in retinal disorders. Ther Adv Ophthalmol. 2021;13:25158414211027708. https://doi.org/10.1177/25158414211027708 (eCollection Jan–Dec 2021).
Gelfman CM, Grishanin R, Bender KO, Nguyen A, Greengard J, Sharma P, Nieves J, Kiss S, Gasmi M. Comprehensive preclinical assessment of ADVM-022, an intravitreal anti-VEGF gene therapy for the treatment of neovascular AMD and diabetic macular edema. J Ocul Pharmacol Ther. 2021;37(3):181–90.
Grishanin R, Vuillemenot B, Sharma P, Keravala A, Greengard J, Gelfman C, Blumenkrantz M, Lawrence M, Hu W, Kiss S, et al. Preclinical evaluation of ADVM-022, a novel gene therapy approach to treating wet age-related macular degeneration. Mol Ther. 2019;27(1):118–29.
Kumar R, Crouthamel MC, Rominger DH, Gontarek RR, Tummino PJ, Levin RA, King AG. Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br J Cancer. 2009;101(10):1717–23.
EyePoint Pharmaceuticals, Inc. EyePoint Pharmaceuticals announces first patient dosed in phase 1 clinical trial of EYP-1901 for the treatment of wet AMD [media release]. 2021. https://www.biospace.com/article/releases/eyepoint-pharmaceuticals-announces-first-patient-dosed-in-phase-1-clinical-trial-of-eyp-1901-for-the-treatment-of-wet-amd/.
Clearside Biomedical, Inc. Clearside Biomedical announces positive safety results from cohort 1 of OASIS phase 1/2a clinical trial of CLS-AX (axitinib injectable suspension) for the treatment of wet AMD [media release]. 2021. https://www.globenewswire.com/news-release/2021/06/15/2247133/0/en/Clearside-Biomedical-Announces-Positive-Safety-Results-from-Cohort-1-of-OASIS-Phase-1-2a-Clinical-Trial-of-CLS-AX-axitinib-injectable-suspension-for-the-Treatment-of-Wet-AMD.html.
Chaney P. Paper presented at Ophthalmology Innovation Summit; San Francisco, CA, USA; 2019.
Wells JA, Gonzales CR, Berger BB, Gonzalez VH, Sippy BD, Burian G. A phase 1, open-label, dose-escalation trial to investigate safety and tolerability of single intravitreous injections of ICON-1 targeting tissue factor in wet AMD. Ophthalmic Surg Lasers Imaging Retina. 2018;49(5):336–45.
Gonzales CR, Burian G. A phase 2 study (EMERGE) evaluating repeated intravitreal administration of ICON-1 in patients with choroidal neovascularization (CNV) secondary to age-related macular degeneration (AMD). Invest Ophthalmol Vis Sci. 2017;58:3766.
Farhat F, Torres A, Park W, de Lima LG, Mudad R, Ikpeazu C, Abi Aad S. The Concept of biosimilars: from characterization to evolution-A narrative review. Oncologist. 2018;23(3):346–52.
Sharma S, Khan M, Chaturvedi A. Group R-ESI: a multicenter, retrospective study (RE-ENACT 2) on the Use of Razumab (World’s First Biosimilar Ranibizumab) in wet age-related macular degeneration. Ophthalmol Ther. 2020;9(1):103–14.
Sharma A, Kumar N, Parachuri N, Bandello F, Kuppermann BD, Loewenstein A. Biosimilars for retinal diseases: an update. Am J Ophthalmol. 2021;224:36–42.
Sharma S, Khan MA, Chaturvedi A. Group R-ESI: real-life clinical effectiveness of razumab(R) (the World’s First Biosimilar of Ranibizumab) in Retinal Vein Occlusion: a subgroup analysis of the pooled retrospective RE-ENACT study. Ophthalmologica. 2019;241(1):24–31.
Griaud F, Winter A, Denefeld B, Lang M, Hensinger H, Straube F, Sackewitz M, Berg M. Identification of multiple serine to asparagine sequence variation sites in an intended copy product of LUCENTIS(R) by mass spectrometry. MAbs. 2017;9(8):1337–48.
Verma L, Thulasidas M, Purohit A, Gupta A, Narula R, Talwar D. Clinical efficacy and safety of Razumab(R) (CESAR) study: Our experience with the world’s first biosimilar Ranibizumab. Indian J Ophthalmol. 2021;69(2):347–51.
Sharma S, Gupta V, Maiti A, Natesh S, Saxena S, Dave V, Parmar V, Sampangi R, Murthy H, Dharwadkar S, et al. Safety and efficacy of Razumab (world’s first biosimilar ranibizumab) in wet age-related macular degeneration: a post-marketing, prospective ASSET study. Int J Retina Vitreous. 2021;7(1):24.
Chakraborty D, Stewart MW, Sheth JU, Sinha TK, Boral S, Das A, Mondal S, Mukherjee A. Real-world safety outcomes of intravitreal ranibizumab biosimilar (razumab) therapy for chorioretinal diseases. Ophthalmol Ther. 2021;10:337–48.
Woo SJ, Veith M, Hamouz J, Ernest J, Zalewski D, Studnicka J, Vajas A, Papp A, Gabor V, Luu J, et al. Efficacy and safety of a proposed ranibizumab biosimilar product vs a reference ranibizumab product for patients with neovascular age-related macular degeneration: a randomized clinical trial. JAMA Ophthalmol. 2021;139(1):68–76.
Taylor P. Europe has its first Lucentis biosimilar, Samsung Bioepis’ Byooviz. 2021. https://pharmaphorum.com/news/europe-has-its-first-lucentis-biosimilar-samsung-bioepis-byooviz/.
Sharma A, Reddy P, Kuppermann BD, Bandello F, Lowenstein A. Biosimilars in ophthalmology: “Is there a big change on the horizon?” Clin Ophthalmol. 2018;12:2137–43.
Sheth JU, Stewart MW, Khatri M, Gupta SR, Chawla S, Rajendran A, Narayanan R. Changing trends in the use of anti-vascular endothelial growth factor (anti-VEGF) biosimilars: Insights from the Vitreoretinal Society of India Biosimilars of Anti-VEGF Survey. Indian J Ophthalmol. 2021;69(2):352–6.
Mirshahi A, Lashay A, Riazi-Esfahani H, Ebrahimiadib N, Khojasteh H, Ghassemi F, Bazvand F, Khodabande A, Roohipour R, Pour EK, et al. Intraocular injection of Stivant (a biosimilar to bevacizumab): a case series. J Ophthalmic Vis Res. 2021;16(1):28–33.
FYB203: aflibercept biosimilar candidate. https://www.formycon.com/en/biosimilars/fyb203/.
Patel S. Medicare spending on anti-vascular endothelial growth factor medications. Ophthalmol Retina. 2018;2(8):785–91.
The L. Age-related macular degeneration: treatment at what cost? Lancet. 2018;392(10153):1090.
Moja L, Lucenteforte E, Kwag KH, Bertele V, Campomori A, Chakravarthy U, D’Amico R, Dickersin K, Kodjikian L, Lindsley K, et al. Systemic safety of bevacizumab versus ranibizumab for neovascular age-related macular degeneration. Cochrane Database Syst Rev. 2014;9:CD011230.
Stewart MW, Narayanan R, Gupta V, Rosenfeld PJ, Martin DF, Chakravarthy U. Counterfeit avastin in India: punish the criminals, not the patients. Am J Ophthalmol. 2016;170:228–31.
Sen S, Mishra C, Kannan NB, Ramasamy K, Rameshkumar G, Lalitha P: Incidence and outcomes of endophthalmitis with in-house compounded intravitreal bevacizumab injections: a multicentric study. Semin Ophthalmol 2021;36(5-6):413–22. https://doi.org/10.1080/08820538.2021.1896746
Kim LN, Mehta H, Barthelmes D, Nguyen V, Gillies MC. Metaanalysis of real-world outcomes of intravitreal ranibizumab for the treatment of neovascular age-related macular degeneration. Retina. 2016;36(8):1418–31.
Kulkarni S, Ramachandran R, Sivaprasad S, Rani PK, Behera UC, Vignesh TP, Chawla G, Agarwal M, Mani SL, Ramasamy K, et al. Impact of treatment of diabetic macular edema on visual impairment in people with diabetes mellitus in India. Indian J Ophthalmol. 2021;69(3):671–6.
Chandra S, Arpa C, Menon D, Khalid H, Hamilton R, Nicholson L, Pal B, Fasolo S, Hykin P, Keane PA, et al. Ten-year outcomes of antivascular endothelial growth factor therapy in neovascular age-related macular degeneration. Eye (Lond). 2020;34(10):1888–96.
Hollingworth W, Jones T, Reeves BC, Peto T. A longitudinal study to assess the frequency and cost of antivascular endothelial therapy, and inequalities in access, in England between 2005 and 2015. BMJ Open. 2017;7(10):e018289.
Yip JLY, Muthy Z, Peto T, Lotery A, Foster PJ, Patel P, Additional UKBE, Vision Consortium M. Socioeconomic risk factors and age-related macular degeneration in the UK Biobank study. BMJ Open Ophthalmol. 2021;6(1):e000585.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The research for this paper was in part financially supported by the Italian Ministry of Health and Fondazione Roma. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
Mariacristina Parravano served on advisory boards for Allergan, Bayer, Novartis, lecture fees from Zeiss and Omikron, honoraria from Alfaintes.
Author contributions
MP: concept and review design; drafting, revision, and final approval of manuscript. EC: review design; drafting and revision of manuscript. GS: review design; revision of manuscript; preparation of tables. GT: review design; revision of manuscript. GV: concept and review design; supervision; drafting, revision, and final approval of manuscript.
Ethics approval
Not applicable.
Consent
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Rights and permissions
About this article

Cite this article
Parravano, M., Costanzo, E., Scondotto, G. et al. Anti-VEGF and Other Novel Therapies for Neovascular Age-Related Macular Degeneration: An Update. BioDrugs 35, 673–692 (2021). https://doi.org/10.1007/s40259-021-00499-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-021-00499-2