
Abstract
Tumor-associated antigens (TAAs) have been identified in many malignant tumors. Within these TAAs are peptide sequences that bind major histocompatibility complex (MHC) class I and class II molecules recognized by T cells triggering antigen-specific CD8+ cytotoxic T-cell and CD4+ T-helper cell responses. Efforts to develop vaccines for breast cancer have been underway for more than 20 years, including peptide and whole inactivated tumor cell vaccines as well as antigen-loaded dendritic cell vaccines. The majority of vaccine trials have used peptides, including single-peptide and multiple-peptide formulations using either MHC class I and class II epitopes in oil-based emulsions alone or in combination with an adjuvant, such as granulocyte-macrophage colony-stimulating factor, and Toll-like receptor agonists. Preclinical research in vitro and in animal models has been aimed at improving vaccine efficacy by identifying more immunogenic peptides and combinations of peptides and adjuvants and cytokine adjuvants that induce stronger immune responses and prolong T-cell memory. Clinical studies investigating the therapeutic potential of active immunization using peptide vaccines has found no serious side effects. In this review, we examine TAA peptide-based vaccination regimens showing promise in breast cancer patients that are also being investigated in clinical trials of safety and efficacy. We also discuss the current limitations in the peptide vaccination field and areas for future development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Breast cancer vaccination has made significant strides with peptide vaccination moving beyond single epitope formulations towards multi-peptide or multi-epitope vaccines and long peptides with improved adjuvants (toll-like receptor agonists and dendritic cell activators) and/or immune checkpoint inhibitors to induce a stronger cellular immune response with a longer-lived memory T-cell pool. |
These cancer vaccines still struggle to find an application as a standard therapy in the continuum of breast cancer care, especially in the adjuvant phase for hormone receptor- and HER2/neu- positive breast cancer for which a plethora of powerful adjuvant-phase therapies exist. Thus, a focus on developing vaccines for more aggressive breast cancer sub-types, like the triple-negative and basal sub-types, is needed. |
There is a need to find more exquisitely breast cancer-specific targets for which the immune system has not developed strong self-tolerance against. In this regard, efforts to identify and develop vaccine approaches against mutated antigens (the tumor ‘mutanome’) and against human endogenous retroviral antigens (HERVs) may be helpful to generate more high-affinity and polyfunctional T-cell responses to prevent disease relapse or treat recurrent metastatic disease. |
1 Introduction
Although there have been significant advances in the treatment of breast cancer, approximately 30 % of patients will experience recurrence of their disease. There is a critical need, therefore, to identify novel therapeutic strategies to augment current treatment regimens. Immunotherapy has shown significant promise in the treatment of other solid tumors, including melanoma and renal cell carcinoma, and there is increasing interest in investigating immunotherapeutic strategies in breast cancer.
The mechanisms of interaction between immune cells and various tumor cells are under investigation. Recent studies have shown that increased numbers of tumor-infiltrating lymphocytes (TILs), especially T cells, are associated with improved prognosis and high response rates to neo-adjuvant chemotherapy in breast cancer patients [1–6]. T lymphocytes recognize antigens presented by antigen-presenting cells (APCs) and attack target cells, like tumor cells, that express these antigens after differentiating into effector cells. In cancer patients, cytotoxic T lymphocytes (CTLs) react against self-antigens and cell lineage antigens that become abnormally over-expressed on cancer cells and are presented as peptides on major histocompatibility complex (MHC) class I and class II molecules by APCs. These primed T cells get activated and undergo cell division and differentiate into effector cells and memory cells, able to migrate to different extents into tumor beds. Cancer cells express antigens that differ from antigens expressed by non-cancerous cells because of mutation or over-expression. Some of these tumor-associated antigens (TAAs) are important for tumor cell survival and growth because they play central roles in protecting cells from apoptosis. TAAs have been proposed as potential targets for anti-tumor immune responses. The induction of humoral and cellular immune responses that further enhance de novo anti-tumor adaptive immunity and/or trigger new responses against TAAs make cancer vaccines an attractive alternative to passive immunotherapies using antibodies or T-cell adoptive cell therapy.
In this article, we briefly review some basic concepts on the functioning of the adaptive immune system related to cancer as well as different cancer vaccine approaches, and then discuss in more detail the application and mechanisms of action of peptide vaccines for breast cancer that have recently been tested in clinical trials. We also discuss combination therapies using peptide vaccination and chemotherapy and future prospects for peptide vaccination for breast cancer as well as the current limitations of this approach and where the field should focus on in the future.
2 Basic Immunology Concepts Related to Cancer Vaccines
2.1 T Cells, Antigen-Presenting Cells, and Major Histocompatibility Complexes
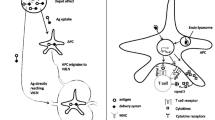
The key adaptive immune system cells that play an important role in peptide vaccination are T cells and APCs. T cells are composed of two main classes, which express CD8 or CD4 co-receptors along with the T-cell receptor (TCR) complex that is the main antigen-recognizing and signaling molecules in T cells. CD4+ T cells, called helper T cells (Th), are mainly divided into Th1 and Th2 subtypes [7, 8], differentiated by the spectrum of cytokines they secrete. Th1 cells produce mainly interleukin (IL)-2, tumor necrosis factor alpha (TNF-α), and gamma interferon (IFN-γ) together with granulocyte-macrophage colony-stimulating factor (GM-CSF) facilitating the activation of dendritic cells (DC) and providing growth-promoting cytokines (IL-2) for CD8+ T cells, while Th2 cells produce mainly IL-4, IL-5, and IL-10 [7, 9]. DCs pick up TAA antigens secreted into the tumor microenvironment or released from exosomes and dead tumor cells and process and present them as peptides on MHC molecules [7, 8]. MHC molecules (also called human leukocyte antigens (HLAs; human MHC) have two forms that bind peptides: class I and class II. MHC class I molecules consist of one chain containing α1 and α2 domains that bind peptides, and a third α3 domain; the alpha chain is associated with a β2-microglobulin molecule that stabilizes this structure. MHC/peptide complexes are recognized by T cells via their TCR. MHC class I peptide complexes are recognized by MHC class I-restricted CD8+ T cells that have been activated and differentiate into cytotoxic effector cells. MHC class I is expressed by all nucleated cells and binds short peptides (8–10 amino acids) [7, 10]. TCR on the surface of the CTL recognize this MHC class I peptide complex in conjunction with the CD8 co-receptor that strengthens or stabilizes the TCR-MHC interaction. This stabilization triggers the TCR and associated CD3 complex to signal via tyrosine kinases and adaptor proteins, thus activating the T cell. MHC class II is normally expressed on DCs, macrophages, and some tumor cells. It binds peptides of 13–25 amino acids [7, 11]. MHC class II peptide complexes are recognized by the TCR and CD4 molecule expressed on CD4+ T cells. Th1 CD4+ T cells are particularly important in activating DCs via CD40-ligand (CD40L) binding to CD40 on the DC, while Th2 cells (through IL-4, IL-5, and IL-10) activate B cells to produce antibodies as well as facilitate immunoglobulin class switching to IgG subtypes during immune responses [7, 8].
2.2 Tumor-Associated Antigens and Cytotoxic T Lymphocyte Responses
The amino acid sequences of naturally processed peptides have fixed, allele-specific positions for MHC binding [12]. The position and the molecular structure of the amino acids that directly affect the ability of a peptide to bind the appropriate MHC molecule constitute the peptide binding motif. T-cell responses to TAA-derived peptides depend both on the binding of the peptide to the MHC molecule and on the affinity of the peptide–MHC complex for TCR [13]. CTLs can lyse tumor cells after recognizing the peptides that have been processed from TAAs and presented on the cell surface. A number of available databases help predict whether a given peptide will interact and bind to an MHC molecule. For example, the ‘SYFPEITHI’ database used an algorithm based on previous publications on T-cell epitopes and MHC ligands [14]. Another database, called the BioInformatics and Molecular Analysis Section (BIMAS) database, used data on predicted half-times of dissociation of peptide binding with HLA class I molecules [15]. Other more recent neural network-based databases are also available, such as netMHC3.4 (http://www.cbs.dtu.dk/services/NetMHC/), that predict possible epitopes binding to a wide range of HLA-A, -B, and -C alleles as well as possible epitopes binding to MHC class II alleles (http://www.cbs.dtu.dk/services/NetMHCII/).
TAAs are predominantly self-proteins expressed at a higher level than in non-cancerous cells. Although central and peripheral tolerance mechanisms in the immune system usually prevent adaptive immune responses by T cells against self-antigens, the increased expression of TAAs can ‘break tolerance’ and facilitate CTL responses to these over-expressed cancer self-antigens as a result of increased CTL-APC avidity. Four types of TAAs have been described (Table 1): (i) overexpressed differentiation antigens arising from the source tissue or cell lineage of the tumor, (ii) Cancer-Testis (CT) antigens expressed normally in germ cells of the testis or ovary, but over-expressed in many cancers, (iii) self-antigens expressed in normal tissues but over-expressed in tumors, and (iv) neo-antigens arising from mutated genes in cancer cells (the so-called ‘mutanome’) [16, 17].
2.3 T-Cell Receptor Signaling, Costimulatory Molecules, and Co-Inhibitory Molecules
Co-stimulatory molecules are essential for TCR signals to generate active CD8+ T cells; however, DCs need to be activated or matured through innate immune signals such as Toll-like receptors (TLRs) to express these critical T-cell co-stimulatory ligands. One of these critical ligands is B7, composed of either CD86 or CD80, which binds and activates the CD28 molecule present on T cells [18]. Other co-stimulatory signaling components expressed on T cells include other immunoglobulin superfamily members such as ICOS (inducible co-stimulator) [19, 20] and members of the TNF receptor superfamily, such as 4-1BB (CD137) and OX40 (CD134) [21, 22]. Matured DCs express the corresponding ligands for these T-cell co-stimulatory molecules. In addition, mature DCs can produce a potent cytokine activating Th1 CD4+ T cells and CD8+ CTL, called IL-12 [7]. Maturation factors for DCs include ligands for TLRs expressed on DCs. These ligands are derived from microbial and viral products and include foreign danger signals such as CpG and free single- and double -stranded RNA, and components of bacterial and fungal cell proteins. These so-called pathogen-associated molecular patterns (PAMPs) bind to TLRs and activate DCs and other APCs through the classical and non-classical NFκB pathway. Components from dead cells and apoptotic bodies (e.g., from dying or dead tumor cells), such as released mitochondrial DNA and heat shock proteins, also bind to innate receptors on DCs and can trigger DC maturation through the activation of type I IFN signaling. Activated Th1 CD4+ T cells are also critical in maturing DCs by expressing CD40-ligand that binds to the CD40 receptor on DCs activating NFκB signaling and thus DC maturation. Type I IFNs, such IFN-α and IFN-β, produced at high levels by activated plasmacytoid DCs (pDCs), are critical in activating natural killer (NK) cells and myeloid-derived DCs to mature into potent APCs [23]. When antigen is presented by immature DCs lacking the expression of the T-cell costimulatory ligands or by non-professional APCs also lacking T-cell costimulatory ligands, a state of T-cell ‘anergy’ can occur in which T cells remain unresponsive for long periods of time, even after later stimulation by mature DCs [23, 24]. This anergy is a major mechanism of self-tolerance in the immune system and can play a role in preventing adequate anti-tumor immune response when antigen is presented by tumor cells (non-professional APCs) to T cells. Without adequate co-stimulation, even T memory cells are only transiently activated and possess low cytotoxic activity [7].
Immunosuppressive tumor microenvironments inhibit local antitumor immune responses by distinct mechanisms, such as impairment of antigen presentation, activation of co-inhibitory signals on T cells such as PD-1 (programmed death-1), active biosynthesis of immunosuppressive molecules, recruitment of naturally occurring regulatory T cells (Tregs), and transformation of effector T cells locally into Tregs [25]. Cytotoxic T-lymphocyte antigen 4 (CTLA-4), which is a member of the immunoglobulin superfamily, inhibits effector T-cell activation by competing with CD28 for CD80/86 binding. CTLA-4, like PD-1, shuts off TCR signaling by activating specific phosphatases (e.g., SHP1 and SHP2), reversing the activating effects of tyrosine kinases [26]. The B7 molecules CD80 and CD86 on APCs have a higher affinity to CTLA-4 (especially CD80) than to CD28 and can trigger CTLA-4 signaling over CD28 costimulation, thereby shutting down T-cell activation [27]. PD-1 binds to PD-l ligand (PD-L1) found on many somatic cells in the body including tumor cells [27]. Blocking CTLA-4 and PD-1 (also called T-cell ‘checkpoints’) has become a powerful new avenue of cancer immunotherapy [26, 28].
3 Cancer Vaccination Strategies
A number of different vaccination strategies have been tested in numerous clinical trials for both metastatic breast cancer and in the adjuvant phase after removal of primary and loco-regional disease in the breast and local lymph nodes. These approaches include short and long single peptide or multi-peptide vaccines [29–31], whole tumor cell vaccines [32, 33], DC-based vaccines [34–37], viral vector vaccines such as pox viruses and adenoviruses encoding TAA genes [38], and oncolytic viral vaccines [38].
3.1 Tumor Cell Vaccines
Tumor cell vaccines consisted of irradiated, freshly isolated primary tumor cells inoculated subcutaneously in an oil-based emulsion such as Incomplete Freund’s Adjuvant (IFA) and other oil-based emulsions or saline [39]. In some cases, tumor cells have been transduced with cytokines such as GM-CSF or viruses such as Newcastle disease virus that triggers a transient and high secretion of IFN-α by the infected cells with the aim of facilitating both NK cell and DC activation at the vaccine site. An interesting clinical trial by Haas and Schirrmacher [40] with high-risk breast cancer patients reported a significant increase in progression-free survival using a whole tumor cell vaccine infected with Newcastle disease virus in the adjuvant phase. This approach and similar whole-cell approaches have been pursued for other solid tumors, including breast cancer, using GM-CSF-transduced cells called GVAXTM [41]. These vaccines have been found to induce cell-mediated immunity against the target cells and TAAs.
3.2 DC-Based Vaccines
DC vaccination usually uses mature DCs that have the highest ability to stimulate T cells, such DCs express both MHC class I and II as well as a host of co-stimulatory ligands, such as CD80, CD86, ICOS-ligand, and ligands for key TNF-R family members such as 4-1BB/CD137 and OX40/CD134. The ability to produce IL-12 is also a key component of these DC vaccines [23]. The most significant disadvantage of autologous DC-based vaccination is that it is too complex to generate reproducibly and in most cases must be customized for each patient [1]. Sipuleucel-T™, the only cancer vaccine currently approved by the FDA, is an example of a DC-enriched vaccine. This vaccine, which is indicated for patients with metastatic castration-resistant prostate cancer, uses a patient’s own peripheral blood mononuclear cells, which include DCs cultured with a proprietary antigen that includes prostatic acid phosphatase and GM-CSF [42]. DC vaccination targeting human epidermal growth factor receptor 2 (HER2) has also been developed in patients with HER2-overexpressing ductal carcinoma in situ. In an ex vivo study, DCs activated in vitro with IFN-γ together with bacterial lipopolysaccharide (LPS) generated antigen-specific T cells that secreted a high level of IFN-γ to the HER2 epitope [43]. In a clinical trial, 5 of 27 patients who were injected with this DC vaccine intra-nodally before surgery had no evidence of remaining tumor, suggesting that DC can, under certain circumstances, elicit strong anti-tumor cell-mediated immune responses [44]. The use of intra-nodal DC immunization is especially relevant in this case because the DCs and antigen are directly placed into a lymphatic environment where T-cell activation occurs and where more highly responsive central memory CD4+ and CD8+ T cells are also found. Most DC vaccines have used subcutaneous or intradermal routes of administration requiring the DCs to migrate to the draining lymph nodes (DLNs) to activate T cells [45, 46]. Many DCs, however, die in the process and only a fraction of the cells arrive at the DLNs. Thus, intra-nodal injection of DC vaccines bypasses this limiting step and promises to increase the efficiency of DC vaccines for breast cancer. This area should be pursued more aggressively in the field.
3.3 Viral Vector Vaccines
Recombinant viral vector vaccines based on pox viruses, such as New York Vaccinia (NYVAC), Modified Virus of Ankara (MVA), and canary pox virus (ALVACTM) engineered to express breast cancer TAAs such as HER2, p53, and MUC1, have also been tested in clinical trials, mainly in metastatic breast cancer patients [47–49]. Although these viral vaccines do elicit fairly strong CD8+ T-cell responses against the encoded TAAs, boosting the antitumor response with these viral vaccines is problematic due to the strong immunity induced against the viral backbone and in some cases the induction of neutralizing antibodies [50]. Heterologous prime-boosting approaches using different viral vectors in sequential order have been used in an attempt to overcome this problem and have helped to induce stronger T-cell responses [51]. However, the overall problem precluding further intense interest in viral vector-based vaccines is that the immune response focuses too much on responding to the viral antigens and relatively weakly to the encoded TAAs. A similar problem exists with adenovirus-based vaccines, although removing some of the highly immunogenic motifs, such as the E1a and E2b genes, have reduced the problem of neutralizing antibody responses after vaccination [52].
3.4 Single- and Multi-Epitope Peptide Vaccines
Peptide vaccines can be efficiently generated with high purity on a large scale and at low cost. However, peptide vaccination is still only weakly immunogenic for eradicating large tumors. If the tumor burden is very high, especially in a patient with metastatic cancer, the efficacy of peptide vaccination has generally been found to be <4 % [53]. Furthermore, peptide vaccines are restricted by HLA type, which means that only HLA-type-matched patients can take these vaccinations. To overcome HLA restriction, a variety of peptides have been generated and optimized to adjust for the large population of patients [54]. To improve the efficacy of peptide vaccination, investigators have also varied peptide lengths and how these peptides are used, such as combinations or different numbers of peptides. Additionally, peptide vaccines are injected with an adjuvant, such as GM-CSF or IFA, as well as TLR agonists such as CpG to heighten their capability [55].
Most peptide vaccine clinical trials have targeted commonly shared over-expressed self-antigens, tissue-specific antigens, and the CT antigen family using both short and long peptide vaccines and multi-epitope peptide vaccines [31, 56]. Short peptides have generally consisted of epitopes presented on commonly expressed HLA-A alleles in the human population such as HLA-A0201 (HLA-A2). Studies with human cancer patients and animal models have also tested the immunogenicity of vaccination using multiple epitopes recognized by T cells. The rationale for this approach is to increase the TAA coverage of the vaccine and prevent the loss of tumor recognition by the immune response due to antigen or epitope loss in tumors caused by genetic and epigenetic modification. Some clinical trials have used a combination of epitopes from different antigens. In a phase I clinical study of DPX-0907, a vaccine including seven peptides from different antigens (Topoisomerase IIα, TACR/ADAM-17, integrin β8 subunit precursor, junction plakoglobin, EDDR1, BAP31, and Abl binding protein C3) with a polynucleotide-based adjuvant, was shown to be safe and antigen-specific immune responses generated IFN-γ [57]. Fourteen clinical trials are currently investigating vaccines using multiple peptides for breast cancer (https://clinicaltrials.gov/). However, regardless of the efficacy of the multi-peptides or long peptides for cancer, these peptide vaccines with multiple boosts can also lead to immunosuppression. In a mouse model, LaCelle et al. [58] showed that multiple vaccinations induced immunosuppressive CD4+ Foxp3+ Tregs. In addition, Leffers et al. [59] showed that a p53 synthetic long-peptide vaccine induced Th2 cytokine response dominating the p53-specific response. Although GM-CSF can induce suppressive myeloid cells and activate endothelial cell precursors when injected systemically [60], most vaccines using localized GM-CSF have not posed a problem in this respect. However, in some cases such as employing GM-CSF as an adjuvant in whole tumor cell vaccines, the use of GM-CSF has been associated with worse prognosis in melanoma patients [61].
4 Target Antigens for Breast Cancer Peptide Vaccines
Several clinical trials have demonstrated induction of immune responses to TAAs in vaccinated breast cancer patients. The antigens investigated include carcinoembryonic antigen (CEA), HER2, mucin 1 (MUC1) (which is hypo-glycosylated in adenocarcinomas) [62, 63], carbohydrate antigens (Tn, TF, sialyl Tn), and p53. Other antigens that have potential for use in a breast cancer vaccine are the CT antigens such as melanoma-associated antigen (MAGE) and New York Breast Cancer-1 (NY-BR-1), as well as the putative universal tumor antigens survivin and human telomerase reverse transcriptase (hTERT) [37, 64]. Targeting these antigens is discussed below.
4.1 HER2 Peptides
HER2 is one of the most widely overexpressed tumor antigens in several cancers. Experimental and clinical studies have shown that HER2 is an immunogenic molecule that can generate antibody responses and activate HER2 peptide-specific CTLs and Th cells [65–69]. Several different HER2 peptides, such as E75, GP2, and AE37, are being investigated as vaccine antigens in adjuvant phase vaccine trials in breast cancer patients with HER2-amplified or HER2 over-expressing primary tumors. Peptides from HER2 are also now being tested in patients without HER2 amplification with slightly over-expressed HER2 (so-called ‘HER2 1+’ by immunohistochemistry), as these patients may also benefit from such a vaccine.
4.1.1 E75 Peptide Vaccine
E75 (HER2 amino acids 369-377: KIFGSLAFL), which is an HLA class I-restricted peptide that stimulates CTLs, has been the most studied of the HER2-derived peptides in clinical trials [70]. The peptide is derived from the extracellular domain of HER2 and is characterized by HLA-A2 and A3 restriction [71]. Murray et al. reported that four vaccinated patients showed E75-specific CTL responses to E75 that correlated with significant increases in IFN-γ secretion [72, 73]. E75 peptide vaccination for stage III/IV breast cancer patients using GM-CSF as an adjuvant induced HER2-specific IFN-γ-producing CD8+ T cells, but this was a short-lived and weak response [74].
Peoples et al. tested E75 plus GM-CSF administered to node-positive and high-risk node-negative breast cancer patients in the adjuvant setting and showed that E75 may reduce recurrences in disease-free and high-risk breast cancer patients [75, 76]. Peripheral blood mononuclear cells showed specific cytotoxicity against HER2-positive target cells [77]. Although HER2 peptide vaccines were mainly intended for use in patients with HER2 over-expressing tumors (HER2 gene amplification), the E75 vaccine with GM-CSF given in an adjuvant setting has been found to induce antigen-specific CD8+ T-cell responses in patients without HER2 gene amplification with intermediate or low levels of HER2 protein expression. Benavides et al. have shown that patients with low levels of HER2 expression (immunohistochemistry staining of 1+ and 2 +) benefited from vaccination [71, 78]. In fact, given the lower HER2 expression in HER2 1+ and 2+ patients, the immune system may not be as tolerant against this antigen and the vaccine may in fact elicit a higher affinity/avidity T-cell response that may be more potent in protecting against tumor recurrence. Disis et al. [79] have shown that E75 and other HER2 extracellular- and intracellular-domain-derived HLA class II peptides in combination with GM-CSF elicited CTL responses in up to 68 % of patients. Other trials have used HLA class II-restricted HER2-derived peptides consisting of 15 amino acids, including the E75 CTL epitope in the same sequence. Using HER2-derived MHC class II helper epitopes containing the E75 MHC class I epitope, Knutson et al. [80] were able to generate long-lasting HER2-specific, IFN-γ-producing CTL responses.
4.1.2 GP2
GP2 is an HLA-A2-restricted peptide derived from the transmembrane domain of HER2 (amino acids 654–662). Although the affinity of GP2 for HLA-A2 is lower than that of E75, preclinical studies suggest that GP2 may be as effective as E75 [81]. Clinical trials testing the GP2 peptide together with GM-CSF as adjuvant are currently ongoing (NCT00524277).
4.1.3 AE37
AE37 is a hybrid peptide containing the MHC class II-restricted AE36 HER2 peptide epitope (amino acids 776–790: GVGSPYVSRLLGICL) linked to the 4-amino-acid (LRMK) moiety from the Invariant chain (Ii) protein; this moiety is called the Ii-Key peptide. The AE36 portion of AE37, derived from the HER2 intracellular domain, binds promiscuously to a large number of different MHC class II subtypes, facilitating broad coverage in the human population. The Ii-Key peptide is thought to stabilize the AE36 peptide loading onto the HLA class II molecule while being shuttled to the cell surface in recycling class II-containing vesicles. This vaccine primarily stimulates CD4+ Th cells. Adjuvant AE37 vaccination in a phase I clinical trial was safe and well tolerated [82, 83]. Interestingly, the AE37 vaccine had potential to induce significantly increased immune responses in patients who did not receive GM-CSF. Furthermore, CD4+ CD25+ FoxP3+ Treg levels, associated with poor prognosis in breast cancer patients, decreased after patients were vaccinated with AE37 [84].
4.2 MUC-1
MUC-1 is a member of the mucin family, heavily glycosylated proteins that are highly expressed at the cell surface in most cancers of epithelial origin. MUC-1 (950-958) is a promising candidate for peptide vaccination of breast cancer patients [63, 85]. Musselli et al. investigated T-cell response in six patients with stage II-III breast cancer after completion of adjuvant therapy and no existence of disease, or stage IV patients who were free from disease or had stable disease. However, immuno-monitoring of immune responses found that only 3 of 24 patients had increased IFN-γ production to the antigen after a 6-day in vitro re-stimulation assay [86].
Tumor cells commonly have defects in the glycosylation pathways mediated in the Golgi network resulting in aberrant glycosylation or hypo-glycosylation yielding novel tumor antigens [87–89]. The sialyl-Tn (STn) epitope from MUC-1 is an example of one of such carbohydrate-based TAA. Theratope™, a synthetic O-linked disaccharide linked to keyhole limpet hemocyanin (KLH) (STn-KLH), was thought to be a strong candidate for a breast cancer vaccine, and phase I and II studies provided supporting evidence in the form of STn antigen-specific T-cell proliferation and peripheral blood lymphocyte lytic activity [90–93]. Two phase II trials showed that patients receiving the STn-KLH vaccine and cyclophosphamide survived significantly longer on average than did the control patients (overall survival, 19.1 vs. 9.2 months; p = 0.001) [93–95]. In a phase III trial, a combination of vaccination with endocrine therapy was well tolerated by patients with metastatic breast cancer. However, no overall benefit in time to disease progression or survival was observed [96, 97]. In contrast, Ibrahim et al. [97] have shown that in metastatic breast cancer patients without aggressive disease who received concurrent hormone therapy, adding STn-KLH resulted in longer time to progression and overall survival in a randomized international phase III trial.
4.3 NY-ESO-1
NY-ESO-1 is a germ cell protein (CT antigen) that is frequently highly expressed in cancer cells but not in non-cancerous cells. In an open-label phase I study including patients with non-small-cell lung cancer, ovarian cancer, melanoma, and breast cancer (two patients), immunization with the NY-ESO-1 p157-165 peptide (SLLMWITQC), in combination with adjuvant CpG synthetic DNA and IFA, induced T-cell responses [98]. Longer NY-ESO-1 peptides have been used to activate CD4+ T-cell responses. For example, an NY-ESO-1-derived pentadecamer epitope (p134–148) induced CD4+ T-cell responses specific to the HLA-DRB1 subtypes *0101, *0301, *0401, and *0701 [99]. In one of four breast cancer patients, CD4+ T cells responded to NY-ESO-1-expressing target cells [99]. The issue with NY-ESO-1, however, is that it is expressed in a minority of breast tumors (2.1 %) [100] and a homolog of NY-ESO-1 (LAGE) containing similar epitopes has been found to stimulate suppressive CD4+ CD25+ Foxp3+ Tregs [101].
4.4 hTERT
hTERT is widely expressed in human cancers and contributes to oncogenesis by preventing telomere erosion and promoting tumor cell immortality [102]. CD8+ T-cell responses de novo to the hTERT I540 peptide (residues 540–548; ILAKFLHWL) have been observed at relatively high frequencies in blood from certain populations of cancer patients [103, 104]. Domchek et al. [105] conducted a clinical trial of an hTERT I540 vaccine with adjuvant GM-CSF for treatment of HLA-A2-positive metastatic breast cancer resistant to conventional cytotoxic therapy. hTERT-specific T cells and CD8+ tumor-infiltrating T lymphocytes were observed in vaccinated patients. Furthermore, the patients with an immune response to hTERT I540 had longer overall survival on average than did the patients without the response.
A phase I trial is currently investigating a vaccination combining hTERT, survivin, daclizumab (humanized anti-human CD25 monoclonal antibody, to transiently deplete Treg cells), and pneumococcal conjugate vaccine (Prevnar; for augmentation of Th-cell immunity) (ClinicalTrials.gov NCT00573495). Results are not yet available.
4.5 Survivin
Survivin is an inhibitor of apoptosis expressed during fetal development. It contains a single Baculovirus IAP (inhibitor of apoptosis) repeat domain and a carboxyl-terminal RING finger [106]. A phase I study found that in patients with metastatic breast cancer, Survivin-2B [80–88] in combination with IFA induced a high level of peptide-specific CTL response after the fourth vaccination [107]. The correlation between Survivin-specific T-cell responses and tumor regression and patient survival has been found in melanoma patients, but results on a similar clinical trial in breast cancer patients are still pending [108].
4.6 Carcinoembryonic Antigen
CEA is a glycoprotein involved in cell adhesion that is normally expressed during fetal development and in adult tissue on normal epithelial cells, such as the colonic lining [109]. Two different HLA-A2-restricted epitopes were used in preclinical vaccine investigations. In a mouse model, immunizing HLA-A2-restricted CEA(691-699) (YMIGMLVGV) peptide significantly prolonged survival on average compared with non-immunized mice, and target cells expressing HLA-A2 and CEA were killed at a significant rate [110, 111]. In another study, T cells from patients immunized with the CEA(571-579) (YLSGADLNL) peptide lysed autologous B cells presenting this peptide [112]. Although CEA as a tumor antigen for cancer immunotherapy has been studied for a long time, it is considered now as a ‘non-starter’ not only due to the high level of immune tolerance against multiple CEA epitopes, but also the potential life-threatening toxicities associated with strong anti-CEA CD8+ CTL responses. Recent clinical trials using the adoptive transfer of CEA-specific T cells (using gene transfer of a high-affinity CEA-specific TCR into activated peripheral blood T cells) in metastatic colorectal cancer patients did induce significant tumor regression in patients, but this was at the cost of immense life-threatening Grade 4 diarrhea caused by the killing of normal colonic epithelial cells expressing CEA (NCT01723306). CEA is an example of a TAA (like HER2 expressed in the heart, especially) also expressed by ‘indispensable’ normal tissues not only subjected to a high degree of self-tolerance, but against which we cannot induce a potent enough anti-cancer response due to the possibility of severe side effects against normal cells. This issue is still a conundrum in the field and will continue to plague the breast cancer vaccine field. Thus, there is still a need to identify ‘real’ breast cancer-specific antigens and it will be difficult to move the field forward without this.
4.7 NY-BR-1
Microarray gene expression screens on primary breast cancer specimens using large panels of normal tissue RNA [113] as specificity controls as well as a process called serological analysis of gene expression (SEREX) [114] have revealed other antigens that may be more specific and yet to be tested in clinical vaccine trials. One of these TAAs that is highly expressed in breast cancer is NY-BR-1, expressed in >80 % of breast tumors [115–117]. NY-BR-1 is one of the most over-expressed breast cancer-specific antigens found to date, especially in ER+ breast cancer [113]. Wang et al. [118] reported that NY-BR-1 peptide-specific CTLs stimulated by DCs pulsed with the p904 peptide (SLSKILDTV) produced IFN-γ and could kill NY-BR-1-expressing breast tumor cells. A phase I trial will be needed to show the efficacy and safety of peptide vaccines derived from NY-BR-1, especially targeting ER+ breast cancer.
4.8 WT1
The Wilms tumor 1 (WT1) gene is overexpressed in leukemia and a variety of solid tumors. The protein encoded by the WT1 gene is necessary for the development of the kidneys and gonads. A phase I trial of an HLA-A2402-restricted WT1 peptide (amino acids 235–243, CMTWNQMNL) and a modified version of this peptide (amino acids 235–243, CYTWNQMNL) found that the vaccination was safe. This WT1 peptide and modified WT1 peptide were tried in metastatic breast cancer patients. In two patients, shrinkage of breast cancer metastases was induced after progression following mastectomy, hormonal therapy and chemotherapy [119, 120].
4.9 p53
Mutations that inactivate p53 proteins or proteins involved in p53 pathways are the most common genetic alterations in human cancers, and mutations in the p53 gene are found in approximately 30 % of breast carcinomas associated with poor prognosis following conventional therapy [121, 122].
CTLs with HLA class I-restricted specificity for wild-type p53 peptides have been observed in peripheral blood spontaneously in some cancer patients and after p53 peptide vaccination [123–125]. However, the levels of CD4+ CD25+ Treg cells can significantly increase after p53 vaccination [126]. A DC-based p53-targeting vaccine consisting of three wild-type and three modified p53-derived HLA-A*0201-binding peptides was used to immunize HLA-A2-positive patients who had progressive metastatic breast cancer after chemotherapy [41, 127]. In eight of the 19 evaluated patients, the disease stabilized. High levels of serum YKL-40 and IL-6 were associated with poor prognoses. YKL-40, also known as chitinase-3-like protein 1, is one of the inflammatory mediators secreted by macrophages and granulocytes and is able to promote tumor growth by up-regulating anti-apoptotic protein levels in the tumor cells [127]. In a phase II trial of the same vaccine in combination with low-dose IL-2, 5 of 22 patients had CTL reactivity to wild-type p53 epitopes [128].
4.10 Combination of CD4+ T-Helper Cell (HLA class II) Epitopes with HLA Class I Epitopes in Breast Cancer Vaccines
CD8+ T cells also need the ‘help’ of CD4+ Th cells to be activated. Longer peptides cannot bind to MHC class I molecules and have to be taken up and processed by DCs [29, 31]. Presentation of both CD8+ and CD4+ T-cell epitopes by DCs improves the killing capacities of both types of T cells [79, 80, 129]. HLA class II peptides prolong epitope presentation in draining lymph nodes and elicit a strong Th cell response to activate immunological memory, resulting in effective clonal expansion and increased IFN-γ production by effector cells [79, 80, 130–133]. Long peptides consisting of both MHC class I and II epitopes require the proteosomal activity of professional APCs like DCs and improve vaccine efficiency by bringing Th cells and CTL precursors together for a more effective immune response [80, 130].
Two clinical trials in breast cancer have tested whether HER2-specific CD8+ T-cell immunity could be elicited by HER2-derived MHC class II helper peptides together with an HLA class I epitope [79, 80]. In one of these trials, 19 HLA-A2-positive patients received a vaccine preparation consisting of one of three HER2 helper epitopes, which contained HLA-A2-binding motifs. After vaccination, peptide-specific T cells were able to lyse tumor cells, suggesting that HER2 MHC class II-binding peptides that contain MHC class I epitopes can induce long-acting HER2-specific, IFN-γ–producing CD8+ T cells [79, 80].
4.11 Epitope Spreading by Single TAA Peptide Vaccines
Another important concept that emerged from peptide-based vaccines for cancer is ‘epitope spreading’ or ‘antigen spreading’ where the initial adaptive immune response against an immunizing antigen (e.g., peptide) facilitates a domino-like effect boosting previous T-cell responses or inducing new T-cell responses against other epitopes in the same TAA and/or against other TAAs not in the original vaccine [54]. This is a critical concept in cancer vaccines and has been also addressed in the breast cancer peptide vaccine field. For example, a clinical trial by Mittendorf et al. showed that the vaccination of patients with the E75 HER2-derived HLA class I-binding peptide with GM-CSF induced a response to another HLA class I-binding HER2 peptide epitope called GP2 as well as a response against an epitope (E41) from another TAA, the folate-binding protein [134, 135]. This suggests the peptide vaccination using short peptide with adjuvant induced not only antigen-specific CD8+ T cells but also both epitope spreading and antigen spreading.
4.12 Combination of Other Treatments with Peptide Vaccination
To increase the power of peptide vaccination in the adjuvant setting, combination treatments could be tested whereby adding other therapies inducing a stronger effect may reduce tumor burden. First, the combination of peptide vaccines and adoptive cell transfer (ACT) should be considered. ACT by peripheral blood lymphocytes or tumor infiltrating lymphocytes involves the selection of autologous lymphocytes with antitumor activity expressing tumor-specific receptor. Vaccinating with peptides from TAAs before or after ACT may increase tumor-specific T cells in patients. Another possible approach is to combine peptide vaccine (especially long multi-epitope vaccines) with oncolytic viruses such as pox viruses, herpes simplex viruses, vesicular stomatitis virus (VSV) and Maraba virus [35, 136, 137]. These oncolytic viruses are usually injected locally directly into accessible tumors (intra-tumoral injection) where tumor cells are more susceptible to viral infection and death, releasing TAAs to prime T-cell responses that can be further boosted by TAA vaccines [35, 136, 137].
Furthermore, combination with targeting co-inhibitory receptor (e.g., CTLA-4, PD-1, PD-L1, LAG3) or co-stimulatory receptor (e.g., ICOS, OX40, CD40) also has the potential to yield better outcomes for patients. For example, in Stage III–IV high-risk melanoma patients receiving a vaccine consisting of three tumor antigen epitopes (gp100, MART-1, and tyrosinase) with adjuvant Montanide ISA 51 and CTLA-4 monoclonal antibody induced strong CD8+ T-cell response, as determined using enzyme-linked immunospot (ELISPOT) assays [138]. In addition, inhibiting other immunosuppressive pathways may also further boost vaccine-specific T-cell responses and epitope and antigen spreading. For example, the use of an inhibitor of the immunoregulatory enzyme, indoleamine 2,3-dioxygenase (IDO), or antibodies to suppressive cytokines such as transforming growth factor beta (TGF-β) can be considered [139, 140]. IDO inhibits T-cell activation and proliferation through tryptophan degradation. TGF-β is a key immunosuppressive cytokine that inhibits the immune response at multiple levels, including preventing DC maturation and limiting T-cell division after activation [139, 140]. Additionally, targeted therapy with tyrosine kinase inhibitors, such as mutant BRAF inhibitor, could provide immuno-modulatory effects on vaccine-induced T-cell responses. BRAF is mutated in some cancers such as melanoma where V600E mutations are quite prevalent and can be targeted by specific inhibitors by binding to the ATP binding pocket [141]. Paradoxically, these specific mutant BRAF inhibitors actually activate immune cells with wild-type BRAF by inducing the formation of BRAF-CRAF dimers [142] and can potentiate anti-tumor CD8+ T-cell responses and ACT [143].
5 Summary and Future Directions
Ample pre-clinical data have demonstrated efficacy of peptide vaccination in generating immune responses that can potentially kill cancer cells. However, their mechanisms of action warrant further investigation. Peptide vaccination and peptide-pulsed DC vaccination may have potential for use as adjuvant therapy to prolong relapse-free survival and overall survival, but the issue of the extent of the T-cell response and the persistence of adequate memory cells relative to the time of relapse (that can be quite long in many primary breast cancer patients) is a huge issue that needs to be addressed. It is not known how long these adjuvant phase vaccines targeting HER2 epitopes, for example, need to be given as prime-boost regimens and whether they can induce truly long-lived central memory T cells. If these vaccines cannot establish long-lived central memory cells, then periodic boosting will always be required. In addition, newer vaccine formulations testing different adjuvants triggering a stronger innate immune response and APC (DC) activation are needed to enhance both CD8+ and CD4+ T-cell immunity and improve the persistence of memory T cells in this context. Some data suggest that vaccination therapy may be beneficial when combined with standard therapies, such as chemotherapy, given sequentially [144, 145]. However, chemotherapy is still a troublesome option when trying to induce or maintain a cellular immune response and establish long-lived memory cells. Careful selection of suitable chemotherapy agents that are compatible in this regard will be needed, and this is largely an empirical exercise requiring costly clinical trials. However, animal models may help. Furthermore, combination therapies with peptide vaccines and immune checkpoint blockade (e.g., anti-CTLA-4 or anti-PD-1), or adoptive T-cell transfer therapy might deliver a stronger anti-tumor response than peptide therapy alone. A big question remains; what is the future of peptide vaccination in breast cancer care, against the current panel of TAAs?
The notion of starting adjuvant phase vaccines after primary tumor removal may also be a troublesome prospect. The reason is that the source of antigen (TAAs) driving the initial T-cell response and maintaining the TAA-specific effector-memory T cells in the first place (the tumor) is removed; original tumor removal paradoxically also severely reduces the antigen load in the system needed to maintain the turnover and survival of these memory cells. Thus, depending on the rate of attrition of the memory T-cell response, adjuvant-phase vaccine regimens initiated after a long time interval, or after the memory response has been largely lost, may be akin to ‘waking up a dead horse’. This conceptual point has not been debated in the field and work needs to be done to determine the longevity of these initial immune responses after removal of the primary tumor in patients to gauge whether it is worth vaccinating patients in the first place with the TAAs in question. Thus, assuring that an adequate level of the pre-existing TAA response still persists, against which the vaccine is formulated, may be a critical biomarker to select patients in this regard.
One of the critical problems plaguing the breast cancer vaccine field, still largely ignored, is that most TAAs being targeted repeatedly in study after study are from over-expressed tissue differentiation antigens like HER2, MUC1, and CEA and others, and are the subjects of strong self-tolerance mechanisms in the immune system. This tolerance occurs because these antigens are expressed on indispensable normal tissues such as the heart, lung, pancreas, kidney, brain, and colon. It is difficult to balance triggering a strong enough cell-mediated immune response against these antigens to really make a difference in protecting patients from metastatic disease and the toxicities that would be associated with such an immune response. Even other antigens such as Survivin are troublesome, as recent studies have shown that triggering a high-affinity T-cell response against Survivin can actually cause activated T cells to kill each other because Survivin is also a critical anti-apoptotic molecule required by T cells [146]. WT-1, p53, hTERT and other similar TAAs also suffer from similar issues. The most pressing problem is the high degree of pre-existing immune tolerance that has eliminated T cells with TCRs having high affinity for peptide epitopes presented from these antigens on self-MHC molecules. This results in only low-affinity T-cell responses being elicited against these TAAs by any vaccine (regardless of the strength of the adjuvant). Thus, even in primary breast cancer patients with no disease, these adjuvant-phase vaccines will mean little in terms of fighting off newly progressing metastatic lesions due to the weak immune reactivity of these T cells. Unfortunately, this is one of the great lessons of the history of peptide vaccination against TAAs over the last few decades.
However, out of the shadows has arisen a new and exciting set of tumor antigens targeting tumor-specific mutations. Recent data suggests that ultimately the potent T-cell responses against breast cancer may ultimately be against mutated antigens or against the tumor mutanome and these can be harnessed now using our existing genetic technologies. In this case, neo-epitopes from proteins derived from mutated tumor genes presented by DCs trigger potent CD4+ and CD8+ T-cell responses that are not restricted by previous central and peripheral self-tolerance mechanisms. The emerging importance of neo-epitope recognition by T cells in cancer has recently been underscored by data from patients receiving autologous tumor-infiltrating lymphocyte (TIL) therapy. In metastatic stage IV melanoma patients, TILs recognizing mutated antigens were found to expand following adoptive transfer and persist in vivo for long periods of time; these mutanome-specific TIL have been associated with complete durable responses in patients (NCT00003895) [147]. Mutanome-specific T-cell responses may also be useful in eradicating metastatic disease in epithelial-derived cancer, as shown in a report on a cholangiocarcinoma patient with widespread metastatic disease who received autologous TIL highly enriched for a patient-specific mutated antigen and underwent durable tumor regression (>75 % decrease in tumor burden) ongoing for more than 2 years as of writing this review [148]. Newer vaccine strategies are now aimed at identifying tumor mutations using whole exome sequencing and mRNA sequencing and predicting epitopes binding to the patient’s own MHC using epitope binding prediction algorithms that are improving as the databases for prediction expand and predictive neural network-based software improves. These peptides are then synthesized and used with adjuvants as a vaccine. Plasmid or viral vaccines incorporating DNA or RNA of single or multiple mutated antigens in tandem could be another approach, especially by inserting these antigens into DCs and using intra-nodal administration to facilitate a strong T-cell response.
Another strategy that is being used in breast cancer and other tumor types is peptide elution from the surface of tumor cells and using this as a source of vaccine [149]. In this case, fresh tumor specimens are used to elute peptide from MHC molecules using a mild acid treatment and these peptides are used in a mix with adjuvants to vaccinate patients. These peptide mixtures would contain epitopes from mutated genes along with epitopes from normal non-mutated gene products. The major problem with this approach, however, is that the mutated epitopes in these eluted mixtures are rare within an ‘ocean’ of much more abundant self-epitopes. Moreover, these normal protein self-epitopes against which strong self-tolerance mechanisms already exist, trigger regulatory T-cell responses that may shut-off the responses against the mutated epitopes through a ‘cross-tolerance’ mechanism.
Another area worth watching out for in terms of breast cancer vaccines is the area of endogenous retroviral antigens, such as human endogenous retroviral antigen-K (HERV-K) [150]. HERV-K and other HERVs are remnants of ancient retroviral infections that entered the human population millions of years ago [151]. These HERVs represent up to 8.3 % of our genomic sequence [151]. The genes from HERV-K are silenced in normal cells due to stop codons, gene insertions, splice sites, and suppression of transcription by transcriptional repressors in normal cells [152]. Thus, under normal circumstances these genes are not expressed and self-tolerance against HERV-K and other HERVs may not exist. DNA instability inducing frameshifts, mutations, deletions, and insertions during tumor initiation and progression can result in the re-expression of full-length HERV-K genes such as the gag and envelope protein. Recently, both CD4+ T-cell-mediated antibody responses as well as CD8+ T-cell responses against HERV-K epitopes have been detected in breast cancer patients [153]. It will be interesting to test whether peptide or recombinant vaccines against these HERVs along with vaccines against mutated gene products will have activity against breast cancer in the future.
Abbreviations
- APCs:
-
Antigen-presenting cells
- CEA:
-
Carcinoembryonic antigen
- CTLs:
-
Cytotoxic T lymphocytes
- DCs:
-
Dendritic cells
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- HLA:
-
Human leukocyte antigen
- hTERT:
-
Human telomerase reverse transcriptase
- IFN-γ:
-
Gamma interferon
- KLH:
-
Keyhole limpet hemocyanin
- MHC:
-
Major histocompatibility complex
- TAAs:
-
Tumor-associated antigens
- TLRs:
-
Toll-like receptors
References
Disis ML. Immune regulation of cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28(29):4531–8.
Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011;8(3):151–60.
Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28(1):105–13.
Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2011;29(15):1949–55.
Lee AH, Gillett CE, Ryder K, Fentiman IS, Miles DW, Millis RR. Different patterns of inflammation and prognosis in invasive carcinoma of the breast. Histopathology. 2006;48(6):692–701.
Rakha EA, Aleskandarany M, El-Sayed ME, Blamey RW, Elston CW, Ellis IO, et al. The prognostic significance of inflammation and medullary histological type in invasive carcinoma of the breast. Eur J Cancer. 2009;45(10):1780–7.
Murphy K, Travers P, Walport M. Immunobiology. New York and London: Garland Science; 2008.
Liew FY. T(H)1 and T(H)2 cells: a historical perspective. Nat Rev Immunol. 2002;2(1):55–60.
Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170(6):2081–95.
Cresswell P, Bangia N, Dick T, Diedrich G. The nature of the MHC class I peptide loading complex. Immunol Rev. 1999;172:21–8.
Sercarz EE, Maverakis E. Mhc-guided processing: binding of large antigen fragments. Nat Rev Immunol. 2003;3(8):621–9.
Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351(6324):290–6.
Bjorkman PJ. MHC restriction in three dimensions: a view of T cell receptor/ligand interactions. Cell. 1997;89(2):167–70.
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3–4):213–9.
Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152(1):163–75.
Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68(3):889–92.
Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–91.
Robson NC, Hoves S, Maraskovsky E, Schnurr M. Presentation of tumour antigens by dendritic cells and challenges faced. Curr Opin Immunol. 2010;22(1):137–44.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, et al. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci USA. 2002;99(18):11790–5.
Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, et al. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS ONE. 2013;8(4):e60031.
Hernandez-Chacon JA, Li Y, Wu RC, Bernatchez C, Wang Y, Weber JS, et al. Costimulation through the CD137/4-1BB pathway protects human melanoma tumor-infiltrating lymphocytes from activation-induced cell death and enhances antitumor effector function. J Immunother. 2011;34(3):236–50.
Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2(3):151–61.
Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193(2):233–8.
Bhardwaj N. Harnessing the immune system to treat cancer. J Clin Investig. 2007;117(5):1130–6.
Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229(1):12–26.
Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1(3):220–8.
Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, et al. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res. 2001;61(17):6451–8.
Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8(5):351–60.
Peoples GE, Holmes JP, Hueman MT, Mittendorf EA, Amin A, Khoo S, et al. Combined clinical trial results of a HER2/neu (E75) vaccine for the prevention of recurrence in high-risk breast cancer patients: U.S. Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14(3):797–803.
Perez SA, von Hofe E, Kallinteris NL, Gritzapis AD, Peoples GE, Papamichail M, et al. A new era in anticancer peptide vaccines. Cancer. 2010;116(9):2071–80.
Elliott RL, Head JF. Adjuvant breast cancer vaccine improves disease specific survival of breast cancer patients with depressed lymphocyte immunity. Surg Oncol. 2013;22(3):172–7.
Chen G, Gupta R, Petrik S, Laiko M, Leatherman JM, Asquith JM, et al. A Feasibility Study of Cyclophosphamide, Trastuzumab, and an Allogeneic GM-CSF-Secreting Breast Tumor Vaccine for HER2+ Metastatic Breast Cancer. Cancer Immunol Res. 2014;2(10):949–61.
Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29(3):372–83.
Milani A, Sangiolo D, Montemurro F, Aglietta M, Valabrega G. Active immunotherapy in HER2 overexpressing breast cancer: current status and future perspectives. Ann Oncol Off J Eur Soc Med Oncol. 2013;24(7):1740–8.
Brossart P, Wirths S, Stuhler G, Reichardt VL, Kanz L, Brugger W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood. 2000;96(9):3102–8.
Curigliano G, Spitaleri G, Pietri E, Rescigno M, de Braud F, Cardillo A, et al. Breast cancer vaccines: a clinical reality or fairy tale? Ann Oncol Off J Eur Soc Med Oncol. 2006;17(5):750–62.
Tai LH, Auer R. Attacking Postoperative Metastases using Perioperative Oncolytic Viruses and Viral Vaccines. Frontiers in oncology. 2014;4:217.
de Gruijl TD, van den Eertwegh AJ, Pinedo HM, Scheper RJ. Whole-cell cancer vaccination: from autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol Immunother. 2008;57(10):1569–77.
Haas C, Schirrmacher V. Immunogenicity increase of autologous tumor cell vaccines by virus infection and attachment of bispecific antibodies. Cancer immunology, immunotherapy : CII. 1996;43(3):190–4.
Svane IM, Pedersen AE, Johnsen HE, Nielsen D, Kamby C, Gaarsdal E, et al. Vaccination with p53-peptide-pulsed dendritic cells, of patients with advanced breast cancer: report from a phase I study. Cancer Immunol Immunother. 2004;53(7):633–41.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New Engl J Med. 2010;363(5):411–22.
Czerniecki BJ, Koski GK, Koldovsky U, Xu S, Cohen PA, Mick R, et al. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res. 2007;67(4):1842–52.
Sharma A, Koldovsky U, Xu S, Mick R, Roses R, Fitzpatrick E, et al. HER-2 pulsed dendritic cell vaccine can eliminate HER-2 expression and impact ductal carcinoma in situ. Cancer. 2012;118(17):4354–62.
Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5(4):296–306.
Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999;59(1):56–8.
Gholami S, Chen CH, Lou E, De Brot M, Fujisawa S, Chen NG, et al. Vaccinia virus GLV-1h153 is effective in treating and preventing metastatic triple-negative breast cancer. Ann Surg. 2012;256(3):437–45.
Scholl S, Squiban P, Bizouarne N, Baudin M, Acres B, Von Mensdorff-Pouilly S, et al. Metastatic breast tumour regression following treatment by a gene-modified vaccinia virus expressing MUC1 and IL-2. J Biomed Biotechnol. 2003;2003(3):194–201.
Schlom J, Kantor J, Abrams S, Tsang KY, Panicali D, Hamilton JM. Strategies for the development of recombinant vaccines for the immunotherapy of breast cancer. Breast Cancer Res Treat. 1996;38(1):27–39.
Rosenberg SA, Zhai Y, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, et al. Immunizing patients with metastatic melanoma using recombinant adenoviruses encoding MART-1 or gp100 melanoma antigens. J Natl Cancer Inst. 1998;90(24):1894–900.
Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J (Sudbury, Mass). 2011;17(5):359–71.
Bellett AJ, Li P, David ET, Mackey EJ, Braithwaite AW, Cutt JR. Control functions of adenovirus transformation region E1A gene products in rat and human cells. Mol Cell Biol. 1985;5(8):1933–9.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15.
Slingluff CL, Jr. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination? Cancer J (Sudbury, Mass). 2011;17(5):343–50.
Klinman DM, Xie H, Little SF, Currie D, Ivins BE. CpG oligonucleotides improve the protective immune response induced by the anthrax vaccination of rhesus macaques. Vaccine. 2004;22(21–22):2881–6.
Yamada A, Sasada T, Noguchi M, Itoh K. Next-generation peptide vaccines for advanced cancer. Cancer Sci. 2013;104(1):15–21.
Berinstein NL, Karkada M, Morse MA, Nemunaitis JJ, Chatta G, Kaufman H, et al. First-in-man application of a novel therapeutic cancer vaccine formulation with the capacity to induce multi-functional T cell responses in ovarian, breast and prostate cancer patients. J Transl Med. 2012;10:156.
LaCelle MG, Jensen SM, Fox BA. Partial CD4 depletion reduces regulatory T cells induced by multiple vaccinations and restores therapeutic efficacy. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15(22):6881–90.
Leffers N, Lambeck AJ, Gooden MJ, Hoogeboom BN, Wolf R, Hamming IE, et al. Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int J Cancer. 2009;125(9):2104–13.
Eggermont AM. Immunostimulation versus immunosuppression after multiple vaccinations: the woes of therapeutic vaccine development. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15(22):6745–7.
Faries MB, Hsueh EC, Ye X, Hoban M, Morton DL. Effect of granulocyte/macrophage colony-stimulating factor on vaccination with an allogeneic whole-cell melanoma vaccine. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15(22):7029–35.
Beatson RE, Taylor-Papadimitriou J, Burchell JM. MUC1 immunotherapy. Immunotherapy. 2010;2(3):305–27.
Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15(17):5323–37.
Emens LA, Jaffee EM. Cancer vaccines: an old idea comes of age. Cancer Biol Ther. 2003;2(4 Suppl 1):S161–8.
Curcio C, Khan AS, Amici A, Spadaro M, Quaglino E, Cavallo F, et al. DNA immunization using constant-current electroporation affords long-term protection from autochthonous mammary carcinomas in cancer-prone transgenic mice. Cancer Gene Ther. 2008;15(2):108–14.
Baxevanis CN, Sotiropoulou PA, Sotiriadou NN, Papamichail M. Immunobiology of HER-2/neu oncoprotein and its potential application in cancer immunotherapy. Cancer Immunol Immunother. 2004;53(3):166–75.
Mamalaki A, Gritzapis AD, Kretsovali A, Belimezi M, Papamatheakis J, Perez SA, et al. In vitro and in vivo antitumor activity of a mouse CTL hybridoma expressing chimeric receptors bearing the single chain Fv from HER-2/neu- specific antibody and the gamma-chain from Fc(epsilon) RI. Cancer Immunol Immunother. 2003;52(8):513–22.
Disis ML, Calenoff E, McLaughlin G, Murphy AE, Chen W, Groner B, et al. Existent T-cell and antibody immunity to HER-2/neu protein in patients with breast cancer. Cancer Res. 1994;54(1):16–20.
Ward RL, Hawkins NJ, Coomber D, Disis ML. Antibody immunity to the HER-2/neu oncogenic protein in patients with colorectal cancer. Hum Immunol. 1999;60(6):510–5.
Fisk B, Blevins TL, Wharton JT, Ioannides CG. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J Exp Med. 1995;181(6):2109–17.
Mittendorf EA, Clifton GT, Holmes JP, Clive KS, Patil R, Benavides LC, et al. Clinical trial results of the HER-2/neu (E75) vaccine to prevent breast cancer recurrence in high-risk patients: from US Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Cancer. 2012;118(10):2594–602.
Murray JL, Gillogly ME, Przepiorka D, Brewer H, Ibrahim NK, Booser DJ, et al. Toxicity, immunogenicity, and induction of E75-specific tumor-lytic CTLs by HER-2 peptide E75 (369-377) combined with granulocyte macrophage colony-stimulating factor in HLA-A2+ patients with metastatic breast and ovarian cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2002;8(11):3407–18.
Zaks TZ, Rosenberg SA. Immunization with a peptide epitope (p369-377) from HER-2/neu leads to peptide-specific cytotoxic T lymphocytes that fail to recognize HER-2/neu+ tumors. Cancer Res. 1998;58(21):4902–8.
Knutson KL, Schiffman K, Cheever MA, Disis ML. Immunization of cancer patients with a HER-2/neu, HLA-A2 peptide, p 369–377, results in short-lived peptide-specific immunity. Clin Cancer Res Off J Am Assoc Cancer Res. 2002;8(5):1014–8.
Holmes JP, Gates JD, Benavides LC, Hueman MT, Carmichael MG, Patil R, et al. Optimal dose and schedule of an HER-2/neu (E75) peptide vaccine to prevent breast cancer recurrence: from US Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Cancer. 2008;113(7):1666–75.
Mittendorf EA, Holmes JP, Ponniah S, Peoples GE. The E75 HER2/neu peptide vaccine. Cancer Immunol Immunother. 2008;57(10):1511–21.
Mittendorf EA, Storrer CE, Foley RJ, Harris K, Jama Y, Shriver CD, et al. Evaluation of the HER2/neu-derived peptide GP2 for use in a peptide-based breast cancer vaccine trial. Cancer. 2006;106(11):2309–17.
Benavides LC, Gates JD, Carmichael MG, Patil R, Holmes JP, Hueman MT, et al. The impact of HER2/neu expression level on response to the E75 vaccine: from U.S. Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15(8):2895–904.
Disis ML, Gooley TA, Rinn K, Davis D, Piepkorn M, Cheever MA, et al. Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide-based vaccines. J Clin Oncol Off J Am Soc Clin Oncol. 2002;20(11):2624–32.
Knutson KL, Schiffman K, Disis ML. Immunization with a HER-2/neu helper peptide vaccine generates HER-2/neu CD8 T-cell immunity in cancer patients. J Clin Investig. 2001;107(4):477–84.
Carmichael MG, Benavides LC, Holmes JP, Gates JD, Mittendorf EA, Ponniah S, et al. Results of the first phase 1 clinical trial of the HER-2/neu peptide (GP2) vaccine in disease-free breast cancer patients: United States Military Cancer Institute Clinical Trials Group Study I-04. Cancer. 2010;116(2):292–301.
Holmes JP, Benavides LC, Gates JD, Carmichael MG, Hueman MT, Mittendorf EA, et al. Results of the first phase I clinical trial of the novel II-key hybrid preventive HER-2/neu peptide (AE37) vaccine. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26(20):3426–33.
Benavides LC, Sears AK, Gates JD, Clifton GT, Clive KS, Carmichael MG, et al. Comparison of different HER2/neu vaccines in adjuvant breast cancer trials: implications for dosing of peptide vaccines. Expert Rev Vaccines. 2011;10(2):201–10.
Gates JD, Clifton GT, Benavides LC, Sears AK, Carmichael MG, Hueman MT, et al. Circulating regulatory T cells (CD4+ CD25+ FOXP3 +) decrease in breast cancer patients after vaccination with a modified MHC class II HER2/neu (AE37) peptide. Vaccine. 2010;28(47):7476–82.
Rentzsch C, Kayser S, Stumm S, Watermann I, Walter S, Stevanovic S, et al. Evaluation of pre-existent immunity in patients with primary breast cancer: molecular and cellular assays to quantify antigen-specific T lymphocytes in peripheral blood mononuclear cells. Clin Cancer Res Off J Am Assoc Cancer Res. 2003;9(12):4376–86.
Musselli C, Ragupathi G, Gilewski T, Panageas KS, Spinat Y, Livingston PO. Reevaluation of the cellular immune response in breast cancer patients vaccinated with MUC1. Int J Cancer. 2002;97(5):660–7.
Brockhausen I. Mucin-type O-glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep. 2006;7(6):599–604.
Chui D, Sellakumar G, Green R, Sutton-Smith M, McQuistan T, Marek K, et al. Genetic remodeling of protein glycosylation in vivo induces autoimmune disease. Proc Natl Acad Sci USA. 2001;98(3):1142–7.
Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. 2001;291(5512):2370–6.
MacLean GD, Reddish MA, Koganty RR, Longenecker BM. Antibodies against mucin-associated sialyl-Tn epitopes correlate with survival of metastatic adenocarcinoma patients undergoing active specific immunotherapy with synthetic STn vaccine. J Immunother Emphas Tumor Immunol Off J Soc Biol Ther. 1996;19(1):59–68.
Reddish MA, MacLean GD, Poppema S, Berg A, Longenecker BM. Pre-immunotherapy serum CA27.29 (MUC-1) mucin level and CD69+ lymphocytes correlate with effects of Theratope sialyl-Tn-KLH cancer vaccine in active specific immunotherapy. Cancer Immun Immunother. 1996;42(5):303–9.
Hadden JW. The immunology and immunotherapy of breast cancer: an update. Int J Immunopharmacol. 1999;21(2):79–101.
Zeichner SB. The failed Theratope vaccine: 10 years later. The Journal of the American Osteopathic Association. 2012;112(8):482–3.
Miles D, Papazisis K. Rationale for the clinical development of STn-KLH (Theratope) and anti-MUC-1 vaccines in breast cancer. Clin Breast Cancer. 2003;3(Suppl 4):S134–8.
MacLean GD, Miles DW, Rubens RD, Reddish MA, Longenecker BM. Enhancing the effect of THERATOPE STn-KLH cancer vaccine in patients with metastatic breast cancer by pretreatment with low-dose intravenous cyclophosphamide. J Immunother Emphas Tumor Immunol Off J Soc Biol Ther. 1996;19(4):309–16.
Miles D, Roche H, Martin M, Perren TJ, Cameron DA, Glaspy J, et al. Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist. 2011;16(8):1092–100.
Ibrahim NK, Murray JL, Zhou D, Mittendorf EA, Sample D, Tautchin M, et al. Survival advantage in patients with metastatic breast cancer receiving endocrine therapy plus sialyl Tn-KLH vaccine: post hoc analysis of a large randomized trial. J Cancer. 2013;4(7):577–84.
Karbach J, Gnjatic S, Bender A, Neumann A, Weidmann E, Yuan J, et al. Tumor-reactive CD8+ T-cell responses after vaccination with NY-ESO-1 peptide, CpG 7909 and Montanide ISA-51: association with survival. Int J Cancer. 2010;126(4):909–18.
Neumann F, Wagner C, Kubuschok B, Stevanovic S, Rammensee HG, Pfreundschuh M. Identification of an antigenic peptide derived from the cancer-testis antigen NY-ESO-1 binding to a broad range of HLA-DR subtypes. Cancer Immunol Immunother. 2004;53(7):589–99.
Theurillat JP, Ingold F, Frei C, Zippelius A, Varga Z, Seifert B, et al. NY-ESO-1 protein expression in primary breast carcinoma and metastases: correlation with CD8+ T-cell and CD79a+ plasmacytic/B-cell infiltration. Int J Cancer. 2007;120(11):2411–7.
Wang HY, Wang RF. Regulatory T cells and cancer. Curr Opin Immunol. 2007;19(2):217–23.
Masutomi K, Yu EY, Khurts S, Ben-Porath I, Currier JL, Metz GB, et al. Telomerase maintains telomere structure in normal human cells. Cell. 2003;114(2):241–53.
Filaci G, Fravega M, Setti M, Traverso P, Millo E, Fenoglio D, et al. Frequency of telomerase-specific CD8+ T lymphocytes in patients with cancer. Blood. 2006;107(4):1505–12.
Gannage M, Abel M, Michallet AS, Delluc S, Lambert M, Giraudier S, et al. Ex vivo characterization of multiepitopic tumor-specific CD8 T cells in patients with chronic myeloid leukemia: implications for vaccine development and adoptive cellular immunotherapy. J Immunol. 2005;174(12):8210–8.
Domchek SM, Recio A, Mick R, Clark CE, Carpenter EL, Fox KR, et al. Telomerase-specific T-cell immunity in breast cancer: effect of vaccination on tumor immunosurveillance. Cancer Res. 2007;67(21):10546–55.
Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3(8):917–21.
Tsuruma T, Iwayama Y, Ohmura T, Katsuramaki T, Hata F, Furuhata T, et al. Clinical and immunological evaluation of anti-apoptosis protein, survivin-derived peptide vaccine in phase I clinical study for patients with advanced or recurrent breast cancer. J Transl Med. 2008;6:24.
Becker JC, Andersen MH, Hofmeister-Muller V, Wobser M, Frey L, Sandig C, et al. Survivin-specific T-cell reactivity correlates with tumor response and patient survival: a phase-II peptide vaccination trial in metastatic melanoma. Cancer Immunol Immunother. 2012;61(11):2091–103.
Kinugasa T, Kuroki M, Yamanaka T, Matsuo Y, Oikawa S, Nakazato H, et al. Non-proteolytic release of carcinoembryonic antigen from normal human colonic epithelial cells cultured in collagen gel. Int J Cancer. 1994;58(1):102–7.
Bhattacharya-Chatterjee M, Saha A, Foon KA, Chatterjee SK. In: Coligan JE et al, editors. Carcinoembryonic antigen transgenic mouse models for immunotherapy and development of cancer vaccines. Current protocols in immunology. Chapter 20:Unit 20.8;2008.
Kawashima I, Hudson SJ, Tsai V, Southwood S, Takesako K, Appella E, et al. The multi-epitope approach for immunotherapy for cancer: identification of several CTL epitopes from various tumor-associated antigens expressed on solid epithelial tumors. Hum Immunol. 1998;59(1):1–14.
Tsang KY, Zaremba S, Nieroda CA, Zhu MZ, Hamilton JM, Schlom J. Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer Inst. 1995;87(13):982–90.
Radvanyi L, Singh-Sandhu D, Gallichan S, Lovitt C, Pedyczak A, Mallo G, et al. The gene associated with trichorhinophalangeal syndrome in humans is overexpressed in breast cancer. Proc Natl Acad Sci USA. 2005;102(31):11005–10.
Jager D, Unkelbach M, Frei C, Bert F, Scanlan MJ, Jager E, et al. Identification of tumor-restricted antigens NY-BR-1, SCP-1, and a new cancer/testis-like antigen NW-BR-3 by serological screening of a testicular library with breast cancer serum. Cancer immunity. 2002;2:5.
Jager D, Karbach J, Pauligk C, Seil I, Frei C, Chen YT, et al. Humoral and cellular immune responses against the breast cancer antigen NY-BR-1: definition of two HLA-A2 restricted peptide epitopes. Cancer Immun. 2005;5:11.
Jager D, Taverna C, Zippelius A, Knuth A. Identification of tumor antigens as potential target antigens for immunotherapy by serological expression cloning. Cancer Immunol Immunother. 2004;53(3):144–7.
Jager D, Stockert E, Gure AO, Scanlan MJ, Karbach J, Jager E, et al. Identification of a tissue-specific putative transcription factor in breast tissue by serological screening of a breast cancer library. Cancer Res. 2001;61(5):2055–61.
Wang W, Epler J, Salazar LG, Riddell SR. Recognition of breast cancer cells by CD8+ cytotoxic T-cell clones specific for NY-BR-1. Cancer Res. 2006;66(13):6826–33.
Morita S, Oka Y, Tsuboi A, Kawakami M, Maruno M, Izumoto S, et al. A phase I/II trial of a WT1 (Wilms’ tumor gene) peptide vaccine in patients with solid malignancy: safety assessment based on the phase I data. Jpn J Clin Oncol. 2006;36(4):231–6.
Oka Y, Tsuboi A, Taguchi T, Osaki T, Kyo T, Nakajima H, et al. Induction of WT1 (Wilms’ tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc Natl Acad Sci USA. 2004;101(38):13885–90.
Soussi T. The p53 tumor suppressor gene: from molecular biology to clinical investigation. Ann New York Acad Sci. 2000;910:121–37; discussion 37–9.
Chang F, Syrjanen S, Syrjanen K. Implications of the p53 tumor-suppressor gene in clinical oncology. J Clin Oncol Off J Am Soc Clin Oncol. 1995;13(4):1009–22.
Petersen TR, Buus S, Brunak S, Nissen MH, Sherman LA, Claesson MH. Identification and design of p53-derived HLA-A2-binding peptides with increased CTL immunogenicity. Scand J Immunol. 2001;53(4):357–64.
Chikamatsu K, Nakano K, Storkus WJ, Appella E, Lotze MT, Whiteside TL, et al. Generation of anti-p53 cytotoxic T lymphocytes from human peripheral blood using autologous dendritic cells. Clin Cancer Res Off J Am Assoc Cancer Res. 1999;5(6):1281–8.
Sung MH, Simon R. Candidate epitope identification using peptide property models: application to cancer immunotherapy. Methods (San Diego, Calif). 2004;34(4):460–7.
Svane IM, Pedersen AE, Nikolajsen K, Zocca MB. Alterations in p53-specific T cells and other lymphocyte subsets in breast cancer patients during vaccination with p53-peptide loaded dendritic cells and low-dose interleukin-2. Vaccine. 2008;26(36):4716–24.
Svane IM, Pedersen AE, Johansen JS, Johnsen HE, Nielsen D, Kamby C, et al. Vaccination with p53 peptide-pulsed dendritic cells is associated with disease stabilization in patients with p53 expressing advanced breast cancer; monitoring of serum YKL-40 and IL-6 as response biomarkers. Cancer Immunol Immunother. 2007;56(9):1485–99.
Pedersen AE, Stryhn A, Justesen S, Harndahl M, Rasmussen S, Donskov F, et al. Wildtype p53-specific antibody and T-cell responses in cancer patients. J Immunother. 2011;34(9):629–40.
Knutson KL, Disis ML. Augmenting T helper cell immunity in cancer. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5(4):365–71.
Zeng G, Li Y, El-Gamil M, Sidney J, Sette A, Wang RF, et al. Generation of NY-ESO-1-specific CD4+ and CD8+ T cells by a single peptide with dual MHC class I and class II specificities: a new strategy for vaccine design. Cancer Res. 2002;62(13):3630–5.
Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, van der Burg SH, Offringa R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur J Immunol. 2008;38(4):1033–42.
Toes RE, Offringa R, Blom RJ, Melief CJ, Kast WM. Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proc Natl Acad Sci USA. 1996;93(15):7855–60.
Toes RE, Blom RJ, Offringa R, Kast WM, Melief CJ. Enhanced tumor outgrowth after peptide vaccination. Functional deletion of tumor-specific CTL induced by peptide vaccination can lead to the inability to reject tumors. J Immunol. 1996;156(10):3911–8.
Mittendorf EA. HER-2/neu peptide breast cancer vaccines: current status and future directions. Breast Dis A Year Book® Q. 2007;17(4):318–20.
Mittendorf EA, Alatrash G, Xiao H, Clifton GT, Murray JL, Peoples GE. Breast cancer vaccines: ongoing National Cancer Institute-registered clinical trials. Expert Rev Vaccines. 2011;10(6):755–74.
Yu B, Zhang Y, Zhan Y, Zha X, Wu Y, Zhang X, et al. Co-expression of herpes simplex virus thymidine kinase and Escherichia coli nitroreductase by an hTERT-driven adenovirus vector in breast cancer cells results in additive anti-tumor effects. Oncol Rep. 2011;26(1):255–64.
Bridle BW, Stephenson KB, Boudreau JE, Koshy S, Kazdhan N, Pullenayegum E, et al. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther J Am Soc Gene Ther. 2010;18(8):1430–9.
Sanderson K, Scotland R, Lee P, Liu D, Groshen S, Snively J, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23(4):741–50.
Iversen TZ, Andersen MH, Svane IM. The targeting of indole-amine 2,3 dioxygenase-mediated immune escape in cancer. Basic Clin Pharmacol Toxicol. 2014 [Epub ahead of print].
Cohn A, Lahn MM, Williams KE, Cleverly AL, Pitou C, Kadam SK, et al. A phase I dose-escalation study to a predefined dose of a transforming growth factor-beta1 monoclonal antibody (TbetaM1) in patients with metastatic cancer. Int J Oncol. 2014;45(6):2221–31.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. New Engl J Med. 2011;364(26):2507–16.
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30.
Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res Off J Am Assoc Cancer Res. 2010;16(24):6040–8.
Okuno K, Sugiura F, Inoue K, Sukegawa Y. Clinical trial of a 7-Peptide cocktail vaccine with oral chemotherapy for patients with metastatic colorectal cancer. Anticancer Res. 2014;34(6):3045–52.
Andersen MH, Junker N, Ellebaek E, Svane IM. Thor Straten P. Therapeutic cancer vaccines in combination with conventional therapy. Journal of biomedicine & biotechnology. 2010;2010:237623.
Leisegang M, Wilde S, Spranger S, Milosevic S, Frankenberger B, Uckert W, et al. MHC-restricted fratricide of human lymphocytes expressing survivin-specific transgenic T cell receptors. J Clin Investig. 2010;120(11):3869–77.
Lu YC, Yao X, Li YF, El-Gamil M, Dudley ME, Yang JC, et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol. 2013;190(12):6034–42.
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–5.
Rammensee HG, Singh-Jasuja H. HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev Vaccines. 2013;12(10):1211–7.
Zhao J, Rycaj K, Geng S, Li M, Plummer JB, Yin B, et al. Expression of Human Endogenous Retrovirus Type K Envelope Protein is a Novel Candidate Prognostic Marker for Human Breast Cancer. Genes Cancer. 2011;2(9):914–22.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921.
Wang-Johanning F, Frost AR, Jian B, Epp L, Lu DW, Johanning GL. Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene. 2003;22(10):1528–35.
Wang-Johanning F, Radvanyi L, Rycaj K, Plummer JB, Yan P, Sastry KJ, et al. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 2008;68(14):5869–77.
Mittendorf EA, Gurney JM, Storrer CE, Shriver CD, Ponniah S, Peoples GE. Vaccination with a HER2/neu peptide induces intra- and inter-antigenic epitope spreading in patients with early stage breast cancer. Surgery. 2006;139(3):407–18.
Sears AK, Perez SA, Clifton GT, Benavides LC, Gates JD, Clive KS, et al. AE37: a novel T-cell-eliciting vaccine for breast cancer. Expert Opin Biol Ther. 2011;11(11):1543–50.
Clive KS, Tyler JA, Clifton GT, Holmes JP, Ponniah S, Peoples GE, et al. The GP2 peptide: a HER2/neu-based breast cancer vaccine. J Surg Oncol. 2012;105(5):452–8.
Knutson KL, Disis ML. Expansion of HER2/neu-specific T cells ex vivo following immunization with a HER2/neu peptide-based vaccine. Clin Breast Cancer. 2001;2(1):73–9.
Peoples GE, Gurney JM, Hueman MT, Woll MM, Ryan GB, Storrer CE, et al. Clinical trial results of a HER2/neu (E75) vaccine to prevent recurrence in high-risk breast cancer patients. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23(30):7536–45.
Gilewski T, Adluri S, Ragupathi G, Zhang S, Yao TJ, Panageas K, et al. Vaccination of high-risk breast cancer patients with mucin-1 (MUC1) keyhole limpet hemocyanin conjugate plus QS-21. Clin Cancer Res Off J Am Assoc Cancer Res. 2000;6(5):1693–701.
Pedersen JW, Blixt O, Bennett EP, Tarp MA, Dar I, Mandel U, et al. Seromic profiling of colorectal cancer patients with novel glycopeptide microarray. Int J Cancer. 2011;128(8):1860–71.
Acknowledgements
We thank Dr. Naoto Ueno (Department of Breast Medical Oncology, MD Anderson Cancer Center, Houston, TX, USA) for critical reading of the manuscript and comments. This study was supported by a 2011 My Oncology Dream Award to Michiko Harao supported by the Japanese Cancer Society and the University of Texas, MD Anderson Global Academic Program.
Conflict of interest
The authors have no conflicts of interest regarding the subject matter of this review.
Author Contributions
Conception and design: MH, EM, LR. Manuscript writing: MH, LR. Responsible for overall content: MH and LR.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Harao, M., Mittendorf, E.A. & Radvanyi, L.G. Peptide-Based Vaccination and Induction of CD8+ T-Cell Responses Against Tumor Antigens in Breast Cancer. BioDrugs 29, 15–30 (2015). https://doi.org/10.1007/s40259-014-0114-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-014-0114-1