
Abstract
Talimogene laherparepvec (T-VEC) is the first agent approved for cancer in the emerging class of oncolytic viral therapies. While T-VEC was approved for the treatment of advanced melanoma in 2015, clinical utilization has been hampered by rapid changes in the therapeutic landscape of melanoma related to advances in both immune checkpoint blockade and targeted therapy, cumbersome logistics involved in T-VEC administration, biosafety concerns, and a perception that T-VEC has limited impact on uninjected, visceral disease. However, with further survival follow-up from the phase III OPTiM (OncovexGM-CSF Pivotal Trial in Melanoma), along with new real-world data and consensus guidelines on safe administration of oncolytic viruses, a roadmap for when and how to use T-VEC has been emerging. In addition, preliminary data have demonstrated improved therapeutic responses to T-VEC in combination with immune checkpoint blockade in patients with melanoma without additive toxicity. This review provides an update on recent data with T-VEC alone and in combination with other agents. The emerging data provide guidance for how to better utilize T-VEC for patients with melanoma and identifies critical areas for clinical investigation to expand the role of T-VEC in combination strategies for the treatment of melanoma and perhaps other cancers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Talimogene laherparepvec (T-VEC) is an approved oncolytic virus for the treatment of melanoma. |
Updated survival data support an overall survival benefit with T-VEC. |
Real-world data suggest the optimal use of T-VEC is in early-stage and first-line settings for melanoma. |
Further studies on combination T-VEC and checkpoint blockade in melanoma are promising. |
1 Introduction
In 2015, the US FDA approved talimogene laherparepvec (Imlygic®; or T-VEC, previously known as OncovexGM-CSF) in the USA for the local treatment of unresectable, cutaneous, subcutaneous, and nodal lesions in patients with recurrence of melanoma following initial surgery, based on results from the randomized phase III open-label OPTiM (Oncovex [granulocyte macrophage colony-stimulating factor (GM-CSF)] Pivotal Trial in Melanoma). The study compared T-VEC with subcutaneous GM-CSF as a control [1]. Since then, the final planned analysis of OPTiM has been published [2]. Compared with GM-CSF, T-VEC resulted in durable responses, improved quality of life, and prolonged survival (unstratified hazard ratio [HR] 0.79 [95% confidence interval {CI} 0.62–1.00]; p = 0.0494 [descriptive]) [2, 3]. The median duration of response among responders was not reached, even with a median follow-up period of over 4 years.
T-VEC is a live, replicating herpes simplex virus, type 1 (HSV-1) that has been modified to express GM-CSF. T-VEC preferentially replicates in tumor cells and has decreased viral pathogenicity by deletion of the ICP34.5 genes that promote neurotoxicity [4]. Through its oncolytic properties, it can release tumor-derived antigens in an immunogenic fashion, produce GM-CSF, and enhance antigen presentation to prime cluster of differentiation (CD)-8+ T-cell responses, which are critical for generating an antitumor immune response [5]. Clinically, T-VEC is administered by direct injection into palpable skin, soft tissue, or nodal melanoma lesions. In cases where tumor is not palpable, ultrasound guidance may be used, but T-VEC has not been approved for visceral tumor injections although ongoing studies are testing the feasibility, safety, and response rates of visceral injection. As currently used, T-VEC indeed appears capable of generating a systemic immune response: a reduction in tumor size of uninjected, non-visceral, and visceral lesions has been observed, although > 50% reduction in tumor size has only been observed in 15% of visceral lesions [1].
The therapeutic efficacy of T-VEC, together with its favorable toxicity profile, makes it an attractive candidate for combination therapy with immune checkpoint inhibitors (ICIs). Three phase Ib/II studies investigating the use of T-VEC in combination with ipilimumab or pembrolizumab have shown very encouraging preliminary results [6,7,8]. These studies have not identified any new adverse events or dose-limiting toxicities associated with combination therapy. We eagerly await the results of an ongoing phase III study investigating T-VEC in combination with pembrolizumab (clinicaltrials.gov: NCT02263508), which completed enrollment of 713 patients and has not yet reported results as prespecified outcomes are event driven. The promising results of these phase Ib/II studies signal that T-VEC could make the treatment relevant for all patients with unresectable, injectable disease who are candidates for ICI, including those with stage IVM1c/b, if results are consistent with those from the early-phase studies.
To date, no other oncolytic viruses have been approved in the USA, and the clinical use of T-VEC has been limited by logistical and biosafety concerns for physicians unfamiliar with local delivery techniques and the approval of many new agents for melanoma, including ICIs and mitogen-activated protein kinase (MAPK)-directed targeted therapy. Nonetheless, emerging real-world data have provided new insights into how best to integrate T-VEC into clinical practice, and several published consensus guidelines have provided advice on how to safely integrate T-VEC into busy ambulatory practice settings. Importantly, there have been no reports of close household contact transmission of T-VEC, suggesting viral transmission is unlikely. Furthermore, updated survival data from OPTiM are now available that support the survival benefit of T-VEC monotherapy [1]. T-VEC is being investigated in combination with other agents and in the neoadjuvant setting for melanoma and several other oncologic indications, including direct delivery to visceral sites of disease. In this review, we discuss the final planned analysis of OPTiM, review the emerging real-world experience using T-VEC across institutions, describe the clinical trial results of T-VEC in combination with ICIs in more detail, review ongoing studies with T-VEC outside of melanoma, and provide practical tips for T-VEC administration.
2 Update on Clinical Trial Results of Talimogene Laherparepvec (T-VEC) in Melanoma
The final planned analysis of overall survival (OS) from OPTiM was performed 3 years after the last patient was randomized (data cut-off 5 September 2014) [2]. In this final analysis, investigators assessed responses and compared them with those reported by the blinded endpoint assessment committee in the primary analysis of OPTiM [1, 2]. Patients with unresectable stage IIIB–IV melanoma according to the 7th edition American Joint Committee on Cancer (AJCC) staging system were eligible if they had one or more cutaneous, subcutaneous, or nodal lesion(s) that was suitable for injection. Patients were allowed to have up to three visceral lesions (excluding lung or nodal lesions associated with visceral organs), measuring no more than 3 cm. Patients were randomized to receive intratumoral T-VEC or subcutaneous GM-CSF. Treatment was administered for 6 months regardless of disease progression unless an alternative therapy was clinically indicated. After 6 months, treatment was stopped in the setting of clinically relevant disease progression, complete remission, lack of response by 12 months, disappearance of injectable lesions, intolerability, or withdrawal of consent. Of the intent-to-treat population (n = 436), 295 patients were assigned to receive T-VEC and 141 to GM-CSF (2:1 randomization). Nearly half of the patients (47%) did not receive prior systemic therapy, although this was allowed.
In the final analysis of OPTiM, the primary endpoint of durable response rate (DRR; rate of complete response [CR] plus partial response lasting at least 6 months) was similar to the primary analysis showing a benefit for T-VEC treatment: 19.3% for T-VEC versus 1.4% for GM-CSF (16.3 vs. 2.1%, respectively, in the primary analysis). Approximately four patients would need to be treated with T-VEC to achieve a DRR relative to GM-CSF by 18 months [3]. Median OS was no different from that reported in the primary analysis: 23.3 months (95% CI 19.5–29.6) with T-VEC and 18.9 months with GM-CSF (95% CI 16.0–23.7). Of patients treated with T-VEC, 16.9% achieved a CR compared with 0.7% of GM-CSF-treated patients. Most importantly, achieving a CR improved OS. The median time to CR for T-VEC was 8.6 months (range 2.1–42.3). Median OS was not reached in patients who achieved a CR, and 72% remained free from recurrence at 3 years following CR, with 88.5% estimated to survive at 5 years.
Multivariate analysis controlling for age, sex, Eastern Cooperative Oncology Group (ECOG) status, line of treatment, and location of melanoma indicated that patients with earlier-stage disease (stage IIIB–IVM1a) and a baseline tumor burden < 14.5 cm2 were more likely to achieve a CR with T-VEC. Among patients receiving T-VEC, the DRR was 33% for stage IIIB/IIIC disease and 16% for stage IVM1a [2]. There was no significant difference between T-VEC or GM-CSF in DRR or overall response rate (ORR) for patients with stage IVM1c. Location of melanoma in the head and neck region was not associated with worse outcomes as may be expected based on prior literature [9]. In this subset, on multivariate analysis, the apparent benefit attributed to location in the head and neck region was no longer significant given the earlier-stage disease in the cohort [2].
Interestingly, 48% of patients who achieved a durable response with T-VEC experienced progression prior to response (PPR) in OPTiM [10]. PPR was defined as the appearance of a new lesion or > 25% increase in baseline total tumor area (the sum of all the products of the two largest perpendicular diameters of all index lesions at baseline) [10]. On a per lesion-level analysis (response of individual injected and uninjected tumoral lesions in 277 patients treated with T-VEC) by the endpoint assessment committee, time to response among responding injected lesions was 9.3 weeks (interquartile range [IQR] 5.1–17.1), with 37% of lesions having a ≥ 50% decrease in total tumor area; 16% completely resolved [10]. Among uninjected, non-visceral lesions, 21% decreased in total tumor area by ≥ 50%, and 14% completely resolved. Responding uninjected lesions were more commonly located on the same body site as injected lesions (48 vs. 23%, respectively). Across patients, most visceral lesions were in the lung, and these were also the most likely visceral lesions to respond to T-VEC. In total, 10% of patients with visceral lesions had a decrease in total tumor area by ≥ 50%; 6% had complete resolution of visceral disease. The median time to response of uninjected non-visceral and visceral lesions was around 12 weeks.
T-VEC has an acceptable safety and tolerability profile [1, 3]. While 99% of patients in OPTiM treated with T-VEC experienced an adverse event (AE), the discontinuation rate because of AEs was 4% [1]. Only 11% experienced a grade 3 or 4 treatment-related AE, the most common of which was injection-site pain or cellulitis [1]. The most common AEs were fatigue (50%), chills (49%), fever (43%), nausea (36%), flu-like illness (30%), and injection-site pain (28%). The incidence of these AEs decreased with subsequent injections. The most common immune-related AE (irAE) was vitiligo, occurring in 6.2%. No grade 4 irAE was noted. Grade 3 irAEs attributed to T-VEC were rare, occurring in patients with predisposing factors, and included glomerulonephritis, lupus vasculitis, pneumonitis, and worsening psoriasis. For patients who responded to T-VEC, 95% experienced an unqualified success (benefit with no harm) [3]. In those who did not respond to T-VEC, 14% experienced an unmitigated failure (harm with no benefit) [3]. In a subgroup analysis of 219 patients with stage IIIB–IVM1a melanoma in OPTiM, significant improvement in 6 of 11 measures (emotional, functional, social/family wellbeing, mental concerns, pain, and ability to work) of the Functional Assessment of Cancer Therapy-Biologic Response Modifier (FACT-BRM) were observed in T-VEC-treated patients compared with those receiving GM-CSF [3].
3 Real-World Data with T-VEC
Since the approval of T-VEC, several centers in the USA and Europe have reported their clinical experience integrating T-VEC into their melanoma treatment paradigm [11,12,13,14,15]. In these cases, the majority of patients treated with T-VEC were stage IIIB–IVM1A according the AJCC 7th edition staging system, which is also the population within OPTiM that experienced the greatest benefit from T-VEC [12, 13, 16]. This is also the currently approved population for T-VEC treatment in Europe. The duration of T-VEC therapy was shorter in these series than in OPTiM. In OPTiM, treatment with T-VEC was continued for a minimum of 6 months per protocol despite evidence of disease progression. Whether patients who discontinued T-VEC because of disease progression would have benefited from ongoing T-VEC administration is unclear. The number of CRs, defined as patients who completed therapy because of an absence of injectable lesions or complete pathologic response, was highly variable among studies, ranging from 11 to 37%. Notable observations, consistent with OPTiM, included an increased duration of therapy in patients who received T-VEC first line and more favorable responses in those with stage IIIB/C disease or a presumably smaller tumor burden given the volume of T-VEC injected. Across series, lesions most commonly injected were cutaneous and located on an extremity.
Of note, several patients within these series received T-VEC concurrent with other systemic therapies, including immunotherapy. Importantly, no new severe AEs were identified in the additional at least 172 patients reported in these series. Novel but non-serious AEs have subsequently been reported and include the development of a granulomatous dermatitis at the site of injection, panniculitis, and Sweet’s-like neutrophilic dermatosis [14, 15, 17]. Granulomatous inflammation may present as inflammatory dermal or subcutaneous papules and, clinically, can mimic lesions of metastatic melanoma; in reported cases, it presented 4–6 months following initiation of T-VEC [14, 15]. Data on the incidence of patients with preexisting autoimmune diseases were not reported. These studies are consistent with the subset analyses in OPTiM, suggesting that T-VEC monotherapy may be better suited for patients with limited disease, such as those with unresectable stage III and IVM1a melanoma and in first-line therapy. Thus, rather than being used as a salvage therapy in treatment-experienced patients, T-VEC monotherapy may be better considered early in the course of melanoma prior to development of widespread systemic metastases.
4 T-VEC Combination Treatment: Emerging Data
ICIs, including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein-1 (PD-1) receptor/ligand inhibitors, enhance T-cell recruitment, prevent exhaustion of activated T cells, and reduce regulatory T-cell (Treg) function. However, their effectiveness may in part depend on the preexistence of an endogenous local antitumor response—a de novo primed antitumor T-cell response. These immunologically “hot” lesions are characterized by the presence of CD8+ tumor-infiltrating lymphocytes (TILs) and an interferon (IFN)-γ inflammatory gene response signature based on evaluation of tumors pretreatment in ICI responders [18]. In addition, programmed death ligand-1 (PD-L1) expression has been a biomarker for response to ICI in certain tumors, such as non-small-cell lung cancer and urothelial tumors, with some evidence of a trend favoring monotherapy with PD-1 blockade in patients with melanoma whose tumors express high levels of PD-L1 [19]. T-VEC, in this respect, becomes an attractive adjunct to checkpoint inhibitors as it can serve to prime the immune system by generating a potent local antitumor immune response by inducing local IFN-γ and PD-L1 expression [23]. In other words, T-VEC could potentially turn immunologically “cold” tumors “hot.”
T-VEC can prime the antitumor immune response via several mechanisms. The antiviral immune response may be a key step in reversing the local immunosuppression in the tumor microenvironment via induction of immunogenic cell death and subsequent release of soluble tumor-associated antigens and danger-associated molecules. Second, infection of the cancer cell results in viral production of human GM-CSF, drawing dendritic cells (DCs) near and promoting their maturation. The release of tumor-derived antigens and viral progeny capable of producing more GM-CSF likely facilitates cross priming of CD8+ T-cell responses by local DCs [5]. The reduction in tumor size of uninjected non-visceral and visceral lesions in OPTiM further supports that T-VEC is producing a systemic antitumor immune response with therapeutic merit beyond its local lytic properties [1]. Based on these mechanistic enhancements in local immune response, the addition of ICIs was anticipated to further expand in situ primed T cells and improve systemic antitumor immunity.
To date, three phase Ib/II studies using T-VEC in combination with the ICIs ipilimumab or pembrolizumab have been published (see Table 1) [6,7,8]. In these studies, patients with unresectable stage IIIB–IVM1c melanoma, excluding those with central nervous system disease, received ICIs starting 6 weeks after a lead-in phase with T-VEC. This design was chosen to avoid rapid T-cell expansion early in T-VEC administration to avoid premature viral clearance by antiviral immune responses. In these trials, study endpoints were assigned per immune-related response criteria (irRC), which allowed for continued treatment, despite apparent growth in existing lesions or development of new lesions, until progression is confirmed no sooner than 4 weeks [20]. The ORR was 39% (CR 13%) and 61.9% (CR 33%) for T-VEC given in combination with ipilimumab or pembrolizumab, respectively [6, 8]. The duration of response was not reached in these studies. These responses are significantly higher than historical controls of these agents when administered as single agents (ipilimumab 11–19%, pembrolizumab 35–40%) for metastatic melanoma [21]. While caution should be used when comparing responses across studies, especially given major differences in eligibility criteria, one of these studies was randomized. In a phase II trial, 198 patients with melanoma were randomized to receive T-VEC with ipilimumab compared with the comparator arm of ipilimumab given alone. Patients who received combination therapy had significantly improved overall responses (combination arm 39%, ipilimumab arm 18%) and decreases in the complete reduction in visceral tumor burden (combination arm 23%, ipilimumab arm 0%). In these studies, no dose-limiting toxicities were identified and no new AEs detected. No subjects discontinued T-VEC because of AEs in the phase Ib studies [7, 8]. T-VEC was discontinued because of AEs in 3 of 98 patients in the phase II randomized study [6]. These data, particularly the responses seen in uninjected visceral disease, strongly support that T-VEC in combination with ipilimumab can generate a greater systemic antitumor response than can ipilimumab alone.
In a phase Ib study evaluating T-VEC in combination with pembrolizumab, the effect of T-VEC on the tumor microenvironment prior to initiation of pembrolizumab was evaluated, with serial biopsies obtained at baseline and on treatment [8]. Prior history of anti-PD-1/PD-L1 therapy, including in the adjuvant setting, was prohibited. As previously mentioned, patients with higher densities of CD8+ T cells, PD-L1 expression, and an IFN-γ gene expression signature are more likely to respond to pembrolizumab [22]. However, in this study, several responders to combination therapy had immunologically “cold” tumors with low levels of CD8+ T-cell infiltration, no PD-L1 expression, or a negative IFN-γ gene signature at baseline. Furthermore, they found that, in 8 of 12 injected lesions available for review, T-VEC increased the density of CD8+ T cells in the injected tumor, and this correlated with response to therapy. Multiplexed immunofluorescence of the tumor microenvironment following T-VEC injections resulted in expansion in CD4+ and CD8+ T cells, many expressing PD-1, and an overall decrease in the Treg to effector T-cell (Teff) ratio. Although this study was small, with only 21 subjects, the overall objective response rate was 62%, and 33% of patients achieved a CR [2]. When taken together, T-VEC could potentially prime the immune system to overcome a primary resistance to pembrolizumab, and combination therapy was well-tolerated with high response rates. The study was expanded into a randomized phase III study investigating T-VEC and pembrolizumab compared with pembrolizumab alone (clinicaltrials.gov: NCT02263508). In this trial, T-VEC was started concurrently with pembrolizumab. The study has completed accrual, and results are expected shortly. Additional studies of T-VEC in melanoma are summarized in Table 2. These include trials evaluating whether T-VEC could reverse an acquired resistance to pembrolizumab; the feasibility and responses of T-VEC in the neoadjuvant setting, prior to definitive surgical resection with or without pembrolizumab or radiation; and T-VEC in combination with the B-Raf proto-oncogene (BRAF)/mitogen-activated protein kinase kinase (MEK) inhibitors dabrafenib and trametinib. Preclinical studies have also suggested synergistic therapeutic responses to a triple combination of T-VEC with MEK inhibition and PD-1 blockade independent of BRAF mutation status [23]. Triple therapy was associated with > 80% survival in murine models of immune-resistant melanoma, and no significant toxicity was noted. However, this concept requires clinical validation [23, 24].
5 T-VEC and Other Indications
T-VEC is also being tested for the treatment of visceral sites of metastatic disease, such as the liver, pancreas, pleura, and peritoneum. Direct injection of hepatocellular carcinoma or liver metastasis of melanoma, breast, or gastrointestinal malignancies is an interesting application of T-VEC under study. The liver is rich in immune cells, but it is normally skewed toward creating a tolerizing immune microenvironment [25]. In melanoma, liver metastasis is associated with an overall poor prognosis and worse responses to ICI [26]. While introducing a live virus into the liver poses unique safety concerns, if T-VEC can help create a more favorable immune microenvironment, in addition to its tumor lytic effect, this could be a significant advance in the treatment of melanoma. However, whether it will be capable of generating as favorable an immune response in the liver as in the skin, where the intradermal generation of antigens has been shown to be an ideal site to generate immunogenic responses and thus serve as a vaccine target organ, remains unknown [27].
6 Integrating T-VEC into Clinical Practice
6.1 Administration and Handling Considerations
T-VEC is the only FDA-approved live, replicating oncolytic virus and, as such, requires special procedures for storage, handling, and administration that cannot readily take advantage of preexisting protocols for drug administration. Individuals who are immunosuppressed or pregnant should not prepare, administer, or come into direct contact with injection sites, dressings, or the bodily fluids of T-VEC-treated patients [28]. Universal biohazard precautions are followed for the handling of T-VEC during its preparation or administration, which includes wearing the following personal protective equipment: protective gown or laboratory coat, safety glasses or face shield, and gloves [28]. Materials that have come in contact with T-VEC must be discarded according to universal biohazard precautions. Patients are advised to discard used dressings and cleaning materials into a sealed plastic bag prior to placing in household waste [28].
T-VEC is provided in two dose strengths, each in single-use 1 mL vials: (1) the 106 (1 million) plaque-forming units (PFU) per mL for the initial dose, and (2) 108 (100 million) PFU per mL for all subsequent doses [28]. Patients receive a low initial dose, which allows HSV-1-naïve patients to seroconvert, which decreases the incidence and severity of potential AEs associated with exposure to higher HSV-1 doses. According to the manufacturer, T-VEC is stored at − 70 °C or colder to prevent thawing, although work is ongoing on a product that will not require − 70 °C storage. T-VEC vials should be thawed in the original carton at room temperature (20–25 °C [68–77 °F]), which can take about 30 minutes. T-VEC should be administered immediately or stored in a refrigerator (2–8 °C [36–46 °F]) in the original vial and carton until later use for up to 24 h (initial dose, 106 PFU per mL vial) or 7 days (108 PFU per mL) or at room temperature (up to 25 °C) for up to 12 h (106 PFU per mL vial) or 24 h (108 PFU per mL vial) [29]. T-VEC must be discarded if not used within this time, which makes it critical that patients be assessed, informed consent obtained, and tumor size measured before T-VEC is thawed so that waste is minimized and the adequate amount needed for injection determined. T-VEC should not be refrozen.
6.2 How to Inject T-VEC
According to the manufacturer, the T-VEC volume to be injected is calculated based on tumor size (longest diameter in cm) of the lesion(s) to be injected (Table 3) [28]. Lesions that are clustered may be considered as one lesion for the purpose of measurement (Fig. 1a). Multiple lesions may be injected up to a maximum of 4 mL of 106 per mL PFU for the initial injection and up to 4 mL of 108 PFU per mL for each subsequent injection. Not all injectable lesions may be injected per session or over the course of treatment given the limitation in total injection volume per visit. For the initial injection, the largest lesion is injected first. For the subsequent sessions, the newest lesion is prioritized, followed by the largest lesions (Fig. 1a). The second visit occurs at 3 weeks after the initial injection to permit seroconversion of HSV-1-negative patients. All subsequent injections occur every 2 weeks for at least 6 months unless other treatment is required or until there are no injectable lesions to treat. If lesions are not easily palpable, portable ultrasound can be used to identify lesions and guide injections.

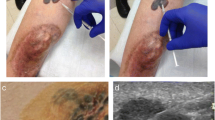
Photograph of a patient with locally recurrent melanoma a before and b after 6 weeks of treatment with talimogene laherparepvec (T-VEC) monotherapy. Three palpable lesions (#1–3, yellow) were selected for injection (prioritized according to size), including two cutaneous (#1 and #3) and one subcutaneous (#2) site. Lesion #1 is a cluster of coalescing papules considered one lesion for the purpose of measuring tumor size (red dashed line indicates the longest tumor diameter). Figure derived from author’s files
Healthcare providers, including nurse practitioners and practice providers, properly trained and supervised by physicians, may inject T-VEC. Preparation of supplies and the patient is key to minimize accidental exposure of staff to T-VEC and to minimize infection risk to the patient. Although no household contact transmission of T-VEC has been reported, inadvertent transmission is possible. T-VEC is sensitive to antiviral medications, such as acyclovir, and sites should have a policy for management of potential needle stick injuries or other exposures. Establishing T-VEC in ambulatory settings may be facilitated by early consultation with hospital epidemiology and infection control committees. This can be an opportunity to provide education on T-VEC, review the safety and viral transmission profile, and remind infection control experts of other live agents, such as Bacillus Calmette Guerin (BCG) used for treatment of noninvasive bladder cancer, that require preparation and transport to cystoscopy facilities and live viral vaccines used in prophylactic disease prevention. In addition, early discussions with local pharmacists who will prepare T-VEC, especially when an oncology-trained pharmacist is available, may help implement T-VEC more easily into clinical practice.
We have found the following procedures best for the administration of T-VEC in our center. Potential patients are evaluated in a dedicated room where they can be seen early for measurement of the melanoma on the day of planned injection. We measure the tumor, then order the drug from the pharmacy while allowing patients a coffee/food break. They are then brought back to the same room for injection when the drug is ready. The same room can be used for multiple patients receiving T-VEC in a given day. When the patient presents, start by measuring the longest tumor diameter of the largest injectable lesion (or start with measuring the newest lesion for subsequent treatment visits) and calculate the total volume of T-VEC needed for the lesion (see Table 3, Fig. 1a). Select additional lesions for possible injection (Fig. 1a). Measure tumor sizes and calculate the volume of T-VEC needed per additional lesion up to a maximum of 4 mL of T-VEC. As the goal is to inject the entire target lesion area using the fewest punctures, we recommend preparing or ordering from the pharmacy, per individual institutional guidelines, syringes of T-VEC in ≤ 1 mL. For example, if target lesion A and B each requires 0.5 mL of T-VEC volume, ideally two syringes, each with 0.5 mL should be prepared. For smaller volumes, insulin syringes in 0.5–1 mL are ideal and should be used with 22- to 26-gauge needles per manufacturer instructions. Depending on the width and depth of the lesion, needle length can be modified.
While syringes of T-VEC are being prepared from frozen stock, prepare the necessary supplies (Table 4). We recommend setting up two surfaces: a sterile and a “dirty” medical instrument tray. At this time, we recommend applying topical anesthetic cream or gel to target lesions (e.g., lidocaine/prilocaine cream [EMLA®] or viscous lidocaine gel) under occlusion for a minimum of 20 min (ideally 1 h) prior to injection. Intralesional short-acting anesthesia (e.g., 1–2% lidocaine) may also be used but should be administered utilizing a ring block closer to the time of T-VEC injection, avoiding infiltration directly into the area to be injected with T-VEC as the anesthetic may affect its efficacy. Alternatively, applying an ice pack over the site of injection for 10–15 min prior to the procedure may also lessen injection-site discomfort. Multiple insertion points may be needed for larger tumors.
Current recommendations are to use universal precautions while handling and injecting T-VEC (see Table 4). We have found it can be helpful to have a prearranged cart with surgical gowns, gloves, face masks, and other T-VEC supplies in the clinic. Prior to injection of T-VEC, place the patient in a comfortable position for maximum access to the injection site, cleanse target lesion(s) with alcohol swabs and allow it to dry. To reduce the risk of infection for the patient, the needle should be changed each time it is removed entirely from the target lesion or between lesions. However, in our experience, for an area consisting of numerous clustered smaller cutaneous target lesions (e.g., individual lesions each < 0.5 cm), using the same syringe and affixed needle is preferable to minimize the risk of needle injury to the healthcare provider.
When ready, inject T-VEC through a single insertion point and distribute the volume across the center of the target lesion, utilizing a fanning technique (or in a “north-south-east-west” manner). Remove needle slowly to exit the lesion and immediately apply 30 s of direct pressure over injection site with a sterile gauze. Clean the injection site with an alcohol swab. Thereafter, change gloves and apply an absorbent dressing (e.g., pad or gauze) and cover with a dry occlusive dressing (e.g., Tegaderm®). Wipe the exterior of the dressing with an alcohol swab. All supplies that have come in contact with T-VEC should be disposed of as biohazard waste per institutional guidance (see Table 4). A systematic evaluation of T-VEC viral shedding and transmissibility found that 100% of patients had T-VEC DNA on the surface of injected lesions and 80% of patients had T-VEC DNA on the exterior of occlusive dressings [29]. However, only 1.1% of swabs taken directly from the surface of injected lesions had the potential for infectivity [29]. T-VEC is sensitive to common viricidal solutions, including 70% isopropyl alcohol and 1% sodium hypochlorite [28]. The table and counters in the room should be cleaned with 10% bleach solution after treatment is completed.
Advise patients to keep the dressing on for at least 1 week, and longer if the injected lesion is oozing [28]. Provide patients with dressings to take home so they can replace the dressing if it falls off. Patients should wear gloves while changing dressings and should avoid touching or scratching injection sites. Prophylactic acetaminophen on the evening of injection may help block the constitutional side effects that may occur 24–48 h after injection. Herpetic infection has been reported in both patients treated with T-VEC and those preparing or administering T-VEC [30]. Individuals with possible herpetic lesions should report the event to Amgen on 1-855-IMLYGIC (1-855-465-9442); patients or close contacts may receive follow-up testing for further characterization of the infection as part of an ongoing study (NCT02910557). T-VEC is sensitive to acyclovir and may be used for healthcare workers or others who have developed a herpetic infection from T-VEC exposure. However, antiviral agents should be used with caution in patients with melanoma as it may interfere with the effectiveness of T-VEC.
7 Conclusion
T-VEC is approved by the FDA and in Europe, Australia, Israel, and Switzerland for the treatment of recurrent unresectable cutaneous, subcutaneous, and/or superficial nodal disease. The favorable toxicity profile, and the potential to generate a systemic antitumor immune response, has led to several clinical trials investigating its use in combination with ICIs in advanced melanoma (stage IIIB or greater). While the results of these phase IB/II studies are encouraging, use of T-VEC in combination with ICI remains under clinical investigation, and a large randomized phase III study of T-VEC and pembrolizumab is expected to report shortly. To date, while T-VEC has been used off-label concurrently with other immunotherapy, consideration of T-VEC is currently recommended for any local satellite, in transit metastasis, or limited to nodal recurrence that is accessible for injection by sight, palpation, or ultrasound. Although surgical resection, if possible, and systemic therapy may also be considered, T-VEC appears to be ideal for patients with limited locoregional disease, when temporal recurrences have become more frequent, and when given as first-line therapy. While patients who are treatment naïve and have a smaller tumor burden (< 14.5 cm2) are more likely to respond to T-VEC, responses have also been seen in those for whom multiple therapies have failed, including targeted therapy and immunotherapy [1, 31]. Further investigation of T-VEC in the neoadjuvant setting and in direct visceral injection of melanoma are also underway, and studies of T-VEC alone and in combination have been initiated in multiple other types of cancer. T-VEC is another arrow in the treatment armamentarium for patients with melanoma and can offer a low toxicity alternative for patients with comorbid conditions that make surgery or systemic therapy more complicated; T-VEC may be especially well-suited for patients with locoregional disease and in the first-line setting. Further clinical studies of T-VEC combinations should help define the role of T-VEC for patients with more advanced systemic melanoma.
References
Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clinl Oncol. 2015;33(25):2780–8. https://doi.org/10.1200/jco.2014.58.3377.
Andtbacka RHI, Collichio F, Harrington KJ, Middleton MR, Downey G, Ӧhrling K, et al. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J Immunother Cancer. 2019;7(1):145. https://doi.org/10.1186/s40425-019-0623-z.
Harrington KJ, Andtbacka RH, Collichio F, Downey G, Chen L, Szabo Z, et al. Efficacy and safety of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in patients with stage IIIB/C and IVM1a melanoma: subanalysis of the Phase III OPTiM trial. Onco Targets Ther. 2016;9:7081–93. https://doi.org/10.2147/OTT.S115245.
Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. https://doi.org/10.1038/sj.gt.3301885.
Dummer R, Hoeller C, Gruter IP, Michielin O. Combining talimogene laherparepvec with immunotherapies in melanoma and other solid tumors. Cancer Immunol Immunother. 2017. https://doi.org/10.1007/s00262-017-1967-1.
Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, open-label Phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36(17):1658–67. https://doi.org/10.1200/JCO.2017.73.7379.
Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34(22):2619–26. https://doi.org/10.1200/JCO.2016.67.1529.
Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170(6):1109-19.e10. https://doi.org/10.1016/j.cell.2017.08.027.
Andtbacka RH, Agarwala SS, Ollila DW, Hallmeyer S, Milhem M, Amatruda T, et al. Cutaneous head and neck melanoma in OPTiM, a randomized phase 3 trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor for the treatment of unresected stage IIIB/IIIC/IV melanoma. Head Neck. 2016;38(12):1752–8. https://doi.org/10.1002/hed.24522.
Andtbacka RH, Ross M, Puzanov I, Milhem M, Collichio F, Delman KA, et al. Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM Phase III clinical trial. Ann Surg Oncol. 2016;23(13):4169–77. https://doi.org/10.1245/s10434-016-5286-0.
Louie KS, Banks V, Scholz F, Richter H, Öhrling K, Mohr P, et al. Real-world use of talimogene laherparepvec in Germany: a retrospective observational study using a prescription database. Futur Oncol. 2020;16(8):317–28. https://doi.org/10.2217/fon-2019-0838.
Mohr P, Haferkamp S, Pinter A, Weishaupt C, Huber MA, Downey G, et al. Real-world use of talimogene laherparepvec in German patients with stage IIIB to IVM1a melanoma: a retrospective chart review and physician survey. Adv Ther. 2019;36(1):101–17. https://doi.org/10.1007/s12325-018-0850-6.
Perez MC, Zager JS, Amatruda T, Conry R, Ariyan C, Desai A, et al. Observational study of talimogene laherparepvec use for melanoma in clinical practice in the United States (COSMUS-1). Melanoma Manag. 2019;6(2):MMT19. https://doi.org/10.2217/mmt-2019-0012.
Lee K, Pouldar D, Shiu J, Elsensohn A, de Feraudy S. The histological spectrum of talimogene laherparepvec (TVEC) injections-Neutrophilic and chronic granulomatous dermatitis. J Cutan Pathol. 2019;46(2):165–7. https://doi.org/10.1111/cup.13387.
Everett AS, Pavlidakey PG, Contreras CM, De Los Santos JF, Kim JY, McKee SB, et al. Chronic granulomatous dermatitis induced by talimogene laherparepvec therapy of melanoma metastases. J Cutan Pathol. 2018;45(1):48–53. https://doi.org/10.1111/cup.13048.
Louie RJ, Perez MC, Jajja MR, Sun J, Collichio F, Delman KA, et al. Real-world outcomes of talimogene laherparepvec therapy: a multi-institutional experience. J Am Coll Surg. 2019;228(4):644–9. https://doi.org/10.1016/j.jamcollsurg.2018.12.027.
Long TH, Shinohara MM, Argenyi ZB, Thompson JA, Gardner JM. Panniculitis in a patient with pathologic complete response to talimogene laherparepvec treatment for recurrent, in-transit melanoma. J Cutan Pathol. 2018;45(11):864–8. https://doi.org/10.1111/cup.13332.
Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17(12):e542–51. https://doi.org/10.1016/s1470-2045(16)30406-5.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. https://doi.org/10.1056/NEJMoa1504030.
Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–20. https://doi.org/10.1158/1078-0432.CCR-09-1624.
Luke JJ. Comprehensive clinical trial data summation for BRAF-MEK inhibition and checkpoint immunotherapy in metastatic melanoma. Oncologist. 2019;24(11):e1197–211. https://doi.org/10.1634/theoncologist.2018-0876.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–40. https://doi.org/10.1172/JCI91190.
Bommareddy PK, Aspromonte S, Zloza A, Rabkin SD, Kaufman HL. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci Transl Med. 2018. https://doi.org/10.1126/scitranslmed.aau0417.
Bommareddy PK, Rabkin SD, Kaufman HL. Triple threat to cancer: rationale for combining oncolytic viruses, MEK inhibitors, and immune checkpoint blockade. Oncoimmunology. 2019;8(4):e1571390. https://doi.org/10.1080/2162402X.2019.1571390.
Crispe IN. Immune tolerance in liver disease. Hepatology. 2014;60(6):2109–17. https://doi.org/10.1002/hep.27254.
Bilen MA, Shabto JM, Martini DJ, Liu Y, Lewis C, Collins H, et al. Sites of metastasis and association with clinical outcome in advanced stage cancer patients treated with immunotherapy. BMC Cancer. 2019;19(1):857. https://doi.org/10.1186/s12885-019-6073-7.
Kupper TS. Old and new: recent innovations in vaccine biology and skin T cells. J Invest Dermatol. 2012;132(3 Pt 2):829–34. https://doi.org/10.1038/jid.2011.400.
BioVex I, a subsidiary of Amgen Inc. IMLYGICTM (talimogene laherparepvec) [package insert]. U.S. Food and Drug Administration website.
Andtbacka RHI, Amatruda T, Nemunaitis J, Zager JS, Walker J, Chesney JA, et al. Biodistribution, shedding, and transmissibility of the oncolytic virus talimogene laherparepvec in patients with melanoma. EBioMedicine. 2019;47:89–97. https://doi.org/10.1016/j.ebiom.2019.07.066.
Soh JM, Galka E, Mercurio MG. Herpetic whitlow-a case of inadvertent inoculation with melanoma viral therapy. JAMA Dermatol. 2018;154(12):1487–8. https://doi.org/10.1001/jamadermatol.2018.3584.
Chesney J, Imbert-Fernandez Y, Telang S, Baum M, Ranjan S, Fraig M, et al. Potential clinical and immunotherapeutic utility of talimogene laherparepvec for patients with melanoma after disease progression on immune checkpoint inhibitors and BRAF inhibitors. Melanoma Res. 2018;28(3):250–5. https://doi.org/10.1097/CMR.0000000000000444.
Author information
Authors and Affiliations
Contributions
CAL conducted research and contributed to writing the manuscript and analysis of the data and provided final approval of the manuscript. NRL contributed to the analysis of the data and editing of the manuscript and provided final approval of the manuscript. AWS contributed to the initial conceptualization of the work, edited the manuscript, helped analyze the data, and provided final approval of the manuscript. HLK provided the initial concept for the manuscript, helped analyze the data and edit the manuscript and provided final approval of the manuscript.
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Howard L. Kaufman is an employee of Immuneering Corporation, serves as a consultant for Replimune, Inc., and is on a clinical advisory board for SapVax. Dr. Cecilia A. Larocca, Dr. Nicole R. LeBoeuf, and Dr. Ann W. Silk have no conflicts of interest that might be relevant to the contents of this manuscript.
Consent for publication
All authors have reviewed the manuscript and agree with revision of this version of manuscript.
Availability of data and material
Any materials or data requested will be made available upon reasonable request.
Rights and permissions
About this article

Cite this article
Larocca, C.A., LeBoeuf, N.R., Silk, A.W. et al. An Update on the Role of Talimogene Laherparepvec (T-VEC) in the Treatment of Melanoma: Best Practices and Future Directions. Am J Clin Dermatol 21, 821–832 (2020). https://doi.org/10.1007/s40257-020-00554-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-020-00554-8