
Abstract
Patients with genetically associated elevated lipoprotein(a) [Lp(a)] levels are at greater risk for coronary artery disease, heart attack, stroke, and peripheral arterial disease. To date, there are no US FDA-approved drug therapies that are designed to target Lp(a) with the goal of lowering the Lp(a) level in patients who have increased risk. The American College of Cardiology (ACC) has provided guidelines on how to use traditional lipid profiles to assess the risk of atherosclerotic cardiovascular disease (ASCVD); however, even with the emergence of statin add-on therapies such as ezetimibe and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, some populations with elevated Lp(a) biomarkers remain at an increased risk for cardiovascular (CV) disease. Residual CV risk has led researchers to inquire about how lowering Lp(a) can be used as a potential preventative therapy in reducing CV events. This review aims to present and discuss the current clinical and scientific evidence pertaining to pelacarsen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pelacarsen’s unique antisense oligonucleotide mechanism allows for the highly specific targeting of apolipoprotein(a) and potent inhibition of lipoprotein(a) [Lp(a)] synthesis. |
Pelacarsen has demonstrated substantial efficacy and safety as an Lp(a)-lowering agent compared with placebo. |
Pelacarsen is the first agent being evaluated for reducing elevated Lp(a) levels and has the potential to be used for the primary and secondary prevention of cardiovascular disease. |
1 Introduction
Cardiovascular (CV) disease (CVD) is the leading cause of death worldwide, accounting for 31% of all global deaths [1]. The relationship between clinical atherosclerotic cardiovascular disease (ASCVD) and prolonged elevated low-density lipoprotein cholesterol (LDL-C) levels has been well-established; however, studies have demonstrated that a residual CV risk remains despite optimal lipid-lowering therapy [2]. A meta-analysis that included seven randomized, controlled, statin outcomes trials showed that patients with established ASCVD and elevated lipoprotein(a) [Lp(a)] levels (> 50 mg/dL) had a 31% greater risk for CVD and an ASCVD event rate of 41.5% despite LDL-C target attainment with statin therapy [2]. There is agreement among a number of expert organizations, including the National Lipid Association (NLA), American Heart Association (AHA), and the American College of Cardiology (ACC), that a level > 50 mg/dL constitutes an ASCVD risk-enhancing factor [3, 4]. In addition, both the European Atherosclerosis Society (EAS) and European Society of Cardiology (ESC) recommend measuring Lp(a) once in each adult’s lifetime to identify those with very high inherited Lp(a) levels (> 180 mg/dL) due to studies showing that Lp(a) levels above 180 mg/dL may be associated with a lifetime risk of ASCVD, equivalent to the risk associated with heterozygous familial hypercholesterolemia (HeFH) [5]. The clinical practice of measuring Lp(a) is not considered standard of care; however, the ACC/AHA guidelines do provide guidance stating a relative indication for its measurement in patients with a family history of premature ASCVD and/or severe hypercholesterolemia (LDL-C >190 mg/dL) [4].
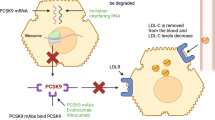
Studies of the proprotein convertase subtilisin/kexin type 9 (PCSK9) monoclonal antibodies evolocumab and alirocumab revealed the Lp(a)-lowering capabilities of these agents, and they were the first agents to show potential CVD benefit with the reduction of Lp(a). A post hoc analysis of the phase III CV outcomes study ODYSSEY showed that alirocumab induced an average 5.0 mg/dL reduction of Lp(a), and that this reduction predicted a 2.5% relative reduction in CV events [3]. In addition, the FOURIER trial showed a similar effect in which evolocumab significantly reduced Lp(a) by 12 mg/dL on average, which corresponded to a 15% decrease in the relative risk of CVD in patients with elevated Lp(a) levels [6, 7]. It should be noted that the extent of Lp(a) lowering observed with alirocumab and evolocumab was dependent on the study population’s baseline Lp(a) levels (initial levels of 58.75 nmol/L and 37 nmol/L, respectively). The mechanism of how PCSK9 inhibitors promote Lp(a) degradation was originally thought to be due to the profound increase in LDL-C receptor (LDLR) expression. This appears to not be the case as a significant discordant response in LDL-C and Lp(a) lowering has been observed with these agents [8]. The lack of correlation between LDL-C and Lp(a) reduction with LDLR expression suggests that PCSK9 inhibition activates alternative mechanisms and additional factors beyond LDLR to determine the extent to which Lp(a) concentrations are lowered. In addition, the degree of Lp(a) lowering has been shown to be variable and may only benefit individuals with very high Lp(a) or those who exhibit larger apolipoprotein(a) [apo(a)] isoforms [8]. The results from PCSK9 studies corroborated past research focused on understanding the role of Lp(a) in CVD.
Elevated Lp(a) levels are present in approximately 20–30% of the general population [9]. Lp(a) is an LDL-like particle, with the apolipoprotein(b) [apo(b)] component covalently bound to apo(a), and is hepatically synthesized (Fig. 1). Lp(a) is highly polymorphic in nature due to the variable number of KIV2 repeats that attribute to the remarkable heritable genetic variation in apo(a) size/mass (small vs. large) seen in the general population [10]. Lp(a) has been discussed with fluctuating highs and lows spanning the past 30 years of atherosclerosis research. Lp(a) first became of interest in the 1980s, then almost succumbed to the trivial research in the 1990s due to flawed epidemiological studies [11]. Novel Mendelian studies with Lp(a) have re-invigorated experts’ quest to investigate the involvement of Lp(a) in CVD [12]. These Mendelian randomization studies supported by genome-wide association studies unraveled the relationship between Lp(a) and atherosclerosis, demonstrating a correlation between genetically inherited Lp(a) levels and CVD [6]. In addition, the independent and causal relationship of Lp(a) with CVD is suggested to correlate at Lp(a) levels above 50 mg/dL [9, 13].

Structure of Lp(a). Lp(a) is composed of apoB-100 covalently bound to apo(a). The highly polymorphic nature of apo(a) is due to the variable number of KIV2 repeats. In this example, the apo(a) isoform has 13 KIV repeats (one copy each of KIV1, KIV3–10 and 4 copies of KIV2). One copy of KV and an inactive protease-like domain are adjacent to the KIV repeats. In addition, oxidized phospholipids (OxPL), are present covalently bound to apo(a) as well as diffused in the lipid phase of apoB-100 [32]. apo(B)-100 apolipoprotein B-100; KIV kringle IV region; Lp(a) lipoprotein(a); OxPL oxidized phospholipids
Oxidized lipids are major factors that cause vascular inflammation in atherosclerosis. A positive association between increased Lp(a) plasma concentrations and calcified aortic valve diseases (CAVD) has been demonstrated in prospective epidemiologic studies [14]. The genetic variation in the Lp(a) locus, which affects Lp(a) concentrations, is known to be linked with aortic valve disease and incident of clinical aortic stenosis [14]. A meta-analysis that included over 7382 CVD cases identified a 2.08-fold increased risk for carriers of small apo(a) isoforms, who, on average, had elevated Lp(a) concentrations [9]. This strong association adds to the probability that Lp(a) is one of the most important genetic risk factors for CVD, given the high frequency of small apo(a) isoforms in the general population [15]. Furthermore, the study also substantiated that this markedly increased CV risk in subjects with elevated Lp(a) involves both proinflammatory and procoagulant effects [9]. Venous thromboembolism (VTE) is a known condition in which pathogenesis involves such systems and there are conflicting data regarding an association of VTE with Lp(a) concentration. Lp(a) lowering with PSCK9 inhibitors have provided insights of this relationship and may mitigate the risk of VTE when added to statin therapy [16]. Many hypotheses exist on the potential of Lp(a) to contribute to CVD through proatherogenic effects of its LDL moiety, proinflammatory effects on its oxidized phospholipid load, and prothrombotic effects through its plasminogen-like protease domain of apo(a).
It is important to note that despite the large body of experimental and clinical evidence supporting the role of Lp(a) in CV risk, the underlying mechanisms responsible for its pathogenicity remain unclear. Contrary to LDL-C, no large outcome studies dedicated to Lp(a) lowering are available, and there are currently no pharmacological-approved therapies that specifically lower Lp(a) concentrations to the extent required to achieve CV benefit, i.e. Lp(a) of approximately ≤ 100 mg/dL [11, 17]. Studies have shown that statin therapy either does not affect or increase Lp(a) levels, while niacin lowers it by approximately 30% but provides no CV benefit [18]. PCKS9 inhibitors have demonstrated Lp(a) lowering; however, this benefit appears to be seen only in patients with very high levels [19]. Additionally, the metabolic pathways of Lp(a) are not fully understood. Studies have demonstrated Lp(a) binding to the LDLR; however, its removal from the plasma via LDLR remains unclear [10]. Other studies have suggested that the differences in Lp(a) plasma concentrations were due to different production rates [10]. The mystery of Lp(a) catabolism has led to a shift in research focused on investigating and developing agents that target Lp(a) assembly. To address this gap in therapy, antisense oligonucleotide (ASO) Lp(a)-targeting agents are in development that have shown reductions in Lp(a) production by up to 80% [20].
1.1 Pelacarsen Molecule and Properties
Pelacarsen is a hepatocyte-directed, second-generation ASO designed to target and bind to apo(a) messenger RNA (mRNA) in hepatocytes, preventing the translation and production of apo(a). Promising data from phase II studies demonstrated Lp(a) lowering of more than 50% with an additional sustained pharmacodynamic (PD) effect and ideal safety profile [21]. The prolonged PD effect allows for monthly dosing, providing benefits of patient convenience and compliance. These results led to the development of the pivotal phase III study Lp(a)HORIZON, which is currently ongoing and aims to confirm the effect of Lp(a) lowering with pelacarsen on CV risk reduction, along with its safety [17, 20].
1.2 Mechanism of Action
Apo(a) is the distinct protein component of Lp(a) primarily responsible for its specific structure and functional properties [22]. Its influence as a key structure and functional component of Lp(a) make it a desirable protein target for interfering with Lp(a) production. With ASO having the capabilities to directly target a protein gene of interest, the ASO platform provides the potential ability to potently reduce Lp(a) levels when administered. Once internalized into the cell via various mechanisms and released into the nucleus, the ASO interacts with its target apo(a) RNA through unique Watson–Crick base-pairing, forming an mRNA-antisense duplex (Fig. 2) [23]. RNase H recognizes the DNA/RNA portion of the duplex through the DNA gap, where it selectively cleaves the apo(a) mRNA molecule for degradation [23]. Afterwards, ASO is liberated and can be recycled, or, in catalytic fashion, seek another apo(a) RNA to complete the total pool of that targeted apo(a) RNA molecule, thus blocking production of apo(a) while subsequently decreasing Lp(a) [20, 23].

The unique ASO mechanism of pelacarsen allows for the highly specific targeting of apo(a) and potent inhibition of Lp(a) synthesis (Fig. 1). To further enhance the pharmacokinetic (PK) and PD profile of pelacarsen, ligand-conjugated antisense (LICA) technology covalently attached to the triantennary N-acetylgalactosamine (GalNAc3) complex was utilized [21]. This unique combination allows for rapid selective uptake via the asialoglycoprotein (ASGP) receptor. The LICA addition showed a more than a 30-fold increase in the potency of the drug [24]. This PK/PD augmentation allows for pelacarsen to be used at lower doses with less-frequent administration compared with non-LICA antisense drugs. Confirmation of the safety and efficacy of pelacarsen in the reduction of Lp(a), along with its tolerability, have been evaluated in phase II clinical studies and are currently being investigated in the phase III pivotal Lp(a)HORIZON study.
2 Clinical Evaluation of Pelacarsen
2.1 Phase I Clinical Studies
A phase I study reported by Tsimikas et al. evaluated the safety, tolerability, PK, and PD of several dosing regimens of pelacarsen in healthy participants with elevated Lp(a), using both a single-dose and multiple-dose study design (Table 1) [25]. The primary outcome studied was percentage change in Lp(a) concentration after 6 months of exposure, while secondary outcomes included percentage changes in Lp(a)-associated oxidized phospholipid content (OxPL-apoB, OxPL-apo(a) OxPL-apoAI, OxPL-PLG), plasminogen, and total cholesterol (TC), along with its components (high-density lipoprotein cholesterol [HDL-C], triglycerides [TGLs], very low-density lipoprotein cholesterol [VLDL-C]). The single-dose study used varying doses of pelacarsen (50, 100, 200, and 400 mg) and included participants aged 18–65 years who had an Lp(a) level of ≥ 25 mg/dL and a body mass index (BMI) of < 32 kg/m2. Participants were randomized (3:1) to either pelacarsen or placebo for a total of five cohorts (three to four participants each) [26]. All participants in the single-dose study completed the study and were included in the safety and full analyses. Overall, no systematic differences in the proportions of participants reporting adverse events (AEs) were observed between treatment groups. No serious AEs (SAEs) were reported, all AEs were of mild severity, and none of the AEs resulted in the discontinuation of study treatment. The most common AE was headache, reported only by participants (n = 2) who had received pelacarsen administered at 200 and 400 mg. In the single-dose phase, pelacarsen administered at 400 mg produced the greatest Lp(a) reduction of 35.9% from baseline at day 30; however, this was not significantly different when compared with placebo. No significant changes were observed in OxPL-apB, OxPL-apo(a), OxPL-apoAI, OxPL-PLG, plasminogen or TC, as well as all cholesterol content with pelacarsen compared with placebo. Following single-dose injections of pelacarsen, median time to maximum plasma concentration (Tmax) ranged from 2 to 4 h after administration. Maximum plasma concentrations (Cmax) were dose-dependent over the analyzed dose range and Cmax was followed by rapid distribution. Results from the single-dose study demonstrated pelacarsen exposure increased in a dose-dependent manner.
The multiple-dose study included three doses of pelacarsen (100, 200, and 300 mg), for a total of six administered doses, in participants aged between 18 and 65 years who met the same criteria as the single-dose previously described [25]. A total of 31 participants were included, with the only significant difference at baseline being BMI between the 100 and 300 mg treatment groups. For all treatment groups at baseline, the mean Lp(a) concentration was 105.0 nmol/L and ranged between 82.2 and 152.3 nmol/L. A loading-dose regimen was used during the first week of treatment (days 1, 3, and 5) to ensure steady-state concentration attainment based on the estimated half-life (t½) of approximately 3 weeks. At day 36, all dosing regimens of pelacarsen resulted in significant decreases in Lp(a) from baseline when compared with placebo (100 mg: − 39.6%, p = 0.005; 200 mg: − 59.0%, p = 0.001; 300 mg: 77.8%, p = 0.001). The safety analysis revealed pelacarsen was generally well-tolerated and AEs were comparable among all groups. The most common AEs were headache (n = 8), fatigue (n = 4), and injection site reactions (n = 10) and all were deemed mild in severity. Pelacarsen administered at 300 mg exhibited the highest incidence of total treatment-emergent AEs (TEAEs) of 41.7%, compared with 12.5% for the 100 mg dose. A total of two participants withdrew from the studies due to AEs, one due to injection site reaction and the other due to a flu-like syndrome that self-resolved with no-long term ramifications. In addition, PK assessment demonstrated total plasma exposure (area under the concentration-time curve from 0 to 24 h [AUC24]) expanded with dose escalation. For both the single- and multi-dose pelacarsen cohorts, TC, apoB, LDL-C, HDL-C, VLDL-C, and TGL levels were not significantly different from placebo following administration. Findings from both phase I studies provide further clinical evidence that supports the ability of pelacarsen to inhibit the synthesis of hepatic-derived target protein [25].
2.2 Phase II Clinical Studies
A randomized, double-blind, dose-finding study evaluated pelacarsen (ranging from 20 to 60 mg) compared with placebo in patients with elevated Lp(a) (≥ 60 mg/dL) [21]. A total of 286 participants with CVD were randomized (5:1) to either pelacarsen or matching placebo using weekly, bi-weekly, or monthly regimens (Table 1). The median age was 60 years and the baseline Lp(a) across all groups ranged from 205 to 247 nmol/L. In addition, the baseline mean LDL-C was 77 mg/dL in the pelacarsen group, compared with 79.4 mg/dL for the placebo group. At trial entry, approximately 80–90% of participants were receiving statin therapy, 50% were receiving ezetimibe, and 20% were receiving PCSK9 inhibitor therapy. The primary endpoint of this study was the percentage change in Lp(a) from baseline to 6 months of exposure.
At day 180, significant dose-dependent reductions in Lp(a) were observed in all pelacarsen cohorts, with decreases of 35%, 56%, 58%, 72%, and 80% (p-value range 0.003 to < 0.001) at doses of 20 mg every 4 weeks, 40 mg every 4 weeks, 20 mg every 2 weeks, 60 mg every 4 weeks, and 20 mg every week, respectively. Notable Lp(a) lowering was observed within the first month of pelacarsen administration and near maximal effect was achieved by week 16. After administration of the last dose, Lp(a) levels returned to baseline within 16 weeks. Further analysis showed that 81% of participants who received pelacarsen 60 mg every 4 weeks and 98% of participants who received 20 mg weekly attained an Lp(a) level of 50 mg/dL or lower (odds ratio [OR] 5.0, 95% confidence interval [CI] 24.0–627.4, and OR 1124.6, 95% CI 109.3–11,571.0, respectively) after 180 days of exposure. Additionally, 71% of participants in the pelacarsen 20 mg weekly cohort achieved an Lp(a) level of 30 mg/dL or lower (OR 347.0, 95% CI 18.3–6597.9). Other relevant markers evaluated were oxidized phospholipid content on both apo(B) and apo(a). After 6 months, pelacarsen resulted in a mean percentage reduction of 37%, 57%, 64%, 79%, and 88% (20 mg every 4 weeks, 40 mg every 4 weeks, 20 mg every 2 weeks, 60 mg every 4 weeks, and 20 mg every week, respectively) in oxidized phospholipids on apo(B), compared with a 14% increase in the placebo group. Apo(a)-oxidized phospholipids were reduced by 28%, 49%, 45%, 63%, and 70% with pelacarsen (20 mg every 4 weeks, 40 mg every 4 weeks, 20 mg every 2 weeks, 60 mg every 4 weeks, and 20 mg every week, respectively), compared with a 20% decrease with placebo.
Pelacarsen appeared to have a relatively benign AE profile, where the incidence of AEs was 90% among participants who received pelacarsen and 83% among participants who received placebo, with injection-site reactions being the most reported AE. Most AEs were deemed to be mild–moderate in severity. In addition, no significant differences between the groups in regard to platelet counts, liver and renal function, or influenza-like symptoms were observed. Pelacarsen administered at 20 mg once weekly and 40 mg every month exhibited the highest incidence of thrombocytopenia (platelet count < 140,000/mm3) (17%, n = 8); however, this observation was comparable with placebo (15%, n = 7). Furthermore, there was no incidence of platelet counts <100,000/mm3 for any treatment groups. Findings from this study showed that pelacarsen significantly reduced Lp(a) levels in patients with CVD in a dose-dependent fashion, and, in addition, revealed favorable safety results regarding thrombocytopenia and ASO therapy by the addition of LICA technology. However, given the relatively small number of participants and the short duration of the study, no definitive conclusions could be drawn regarding the long-term adverse effect profile of pelacarsen .
2.3 Phase III Clinical Studies
To address the limitations of the phase II study, the pivotal phase III L(p)a HORIZON study will be assessing the impact of Lp(a) lowering with pelacarsen on major cardiovascular events (MACE) in patients with established CVD. This long-term CV outcomes study will include approximately 7680 participants aged 18–80 years with established CVD and elevated Lp(a) levels (≥ 70 mg/dL), and is anticipated to conclude in early 2024 (Table 1) [27]. The co-primary endpoints for this study are time to first occurrence of MACE, defined as CV death, non-fatal myocardial infarction (MI), non-fatal stroke, and urgent coronary revascularization requiring hospitalization, with Lp(a) levels ≥70 mg/dL and ≥ 90 mg/dL [27]. Secondary objectives include time to first occurrence of MACE composite endpoint (CV death, non-fatal MI, and non-fatal stroke), time to first occurrence of coronary heart disease (CHD; CHD death, non-fatal MI, and urgent coronary revascularization requiring hospitalization), and number of participants with confirmed all-cause death. The results generated from this study will provide essential evidence to determine whether the marked Lp(a) reductions attained with pelacarsen are transferable into a reduction of CV risk and MACE. It may also bring clarity to the question of whether the strategy of targeting Lp(a) will have beneficial effects on outcomes in patients with CVD whose LDL-C levels are optimally controlled with lipid-lowering therapy.
3 Discussion
Lp(a) has emerged as a promising therapeutic target for CVD. By blocking apo(a) production, significant reductions in Lp(a) levels have been obtained that may lead to a reduction of CV risk. In the phase II study, pelacarsen reduced plasma Lp(a) by more than 50% and this effect was sustained up to 16 weeks upon administration of the last dose [21].
As more research has been directed towards understanding the role of Lp(a) in CV risk, it should be noted that pelacarsen is not the only Lp(a)-lowering agent currently under development. Olpasiran (AMG860) is an Lp(a)-targeting agent that utilizes small-interfering RNA (siRNA) technology and appears to exhibit comparable Lp(a)-lowering effects [28]. A phase I study evaluated the safety, tolerability, PK, and PD effects of olpasiran single-dose treatment in 64 participants who had an Lp(a) level of between 70 and 199 nmol/L. Olpasiran significantly reduced Lp(a), with median percentage reductions of > 90% at doses of ≥ 9 mg and effects persisting for more than 6 months. In addition, olpasiran was well-tolerated, with no safety concerns identified [28].
Although data strongly suggest the potential benefits of lowering elevated Lp(a) levels, the degree of lowering needed to provide clinically meaningful reductions in MACE remains ill-defined. A small study conducted by Stiekema et al. compared the effects of pelacarsen with evolocumab on the activation of proinflammatory monocytes in patients with elevated Lp(a) with or without CVD [29]. Using transcriptome analysis, potent Lp(a) lowering following pelacarsen was observed. Modest Lp(a) lowering combined with LDL-C reduction following evolocumab treatment reduced the proinflammatory state of circulating monocytes [29]. However, the big question remains, is the strategy of potent lowering of Lp(a) compared with moderate lowering seen with PCSK9 inhibitors necessary and will this translate into a CV risk reduction that is seen in the relationship between LDL-C reduction and CVD.
The strategy of potent lowering is supported by multiple studies. One large Mendelian randomization analysis that included over 80,000 patients and more than 150,000 controls evaluated the correlation between lowering LDL-C and Lp(a) with CV risk [30]. The study estimated that an absolute Lp(a) concentration change of 101.5 mg/dL was needed to achieve a relative risk reduction (22–25%) in CHD similar to a reduction of LDL-C by 38.67 mg/dL [30]. These results were supported by another study conducted by Madsen et al. that concluded short-term (i.e. 5 years) lowering of Lp(a) by 50 mg/dL in patients with elevated Lp(a) levels (> 50 mg/dL) may reduce CVD by 20% in the secondary prevention setting [31].
Data from the L(p)a HORIZON study should provide the necessary information to conclude the clinical utility of pelacarsen in patients with elevated Lp(a). In addition, the extended duration of this study may provide insight into the clinical implications on whether obtaining considerably lower Lp(a) levels (< 30 mg/dL) for a prolonged duration provides any further benefit, or, on the contrary, incurs any unfavorable events.
4 Conclusion
Pelacarsen has demonstrated substantial efficacy and safety, when compared with placebo, as an Lp(a)-lowering agent. Pelacarsen is the first agent being evaluated for reducing elevated Lp(a) levels, and has the potential to be used for the primary and secondary prevention of CVD. Pelacarsen provides a novel mechanism with high-specificity that may lower the residual CV risk in patients whose LDL-C is optimally controlled.
Change history
11 December 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40256-021-00513-6
References
World Health Organization. WHO; 2021. https://www.who.int/cardiovascular_disease/world-heart-day/en/. Accessed 22 Apr 2021.
Willeit P, Ridker PM, Nestel PJ, et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet. 2018;392(10155):1311–20.
Wilson DP, Jacobson TA, Jones PE, et al. Use of Lipoprotein(a) in a clinical practice: a biomarker whose time has come A scientific statement from the National Lipid Association. J Cli Lipidol. 2019;13:374–92.
Arnett DK, Blumenthal RS, Albert A, Buroker AB, Goldberger ZD. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease. J Am Coll Cardiol. 2019;74(10):e177-232.
Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risl: The Task Force for the managment of dyslipideaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur Heart J. 2020;41(1):111–88.
O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I. Lipoprotein(a), pcsk9 inhibition, and cardiovascular risk insights from the FOURIER Trial. Circulation. 2019;139:1483–92.
Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular Disease. N Engl J Med. 2017;376(18):1713–22.
Fazio S, Edmiston JB, Brooks N, et al. Discordant response of low-density lipoprotein cholesterol and lipoprotein(a) levels to monoclonal antibodies targeting proprotein convertase subtilisin/kexin type 9. J Clin Lipidol. 2017;11(3):667–73.
Steikema LCA, Prange KHM, Hoogeveen RM, et al. Potent lipoprotein(a) lowering follwing apolipoprotein(a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipoprotein(a). Eur Heart J. 2020;41:2262–71.
Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein(a). J Lipid Res. 2016;57:1339–59.
Coassin S, Kronenberg F. Mechanistic insight into lipoprotein(a): from infamous to “inflammous.” Eur Heart J. 2020;420(41):2272–4.
Kronenberg K, Utermann G. Lipoprotein(a): resurrected by genetics. J Intern Med. 2013;273(1):6–30.
Lamina C, Kronenberg F. Therapeutic effect size of reduction in coronary heart disease outcomes. A Mendelian Randomization Analysis. JAMA Cardiol. 2019;4(6):575–9.
Thanassoulis G. Lipoprotien(a) in calcific aortic valve disease: from genomics to novel drug target for aortic stenosis. J Lipid Res. 2016;57:917–24.
Erqou S, Thompson A, Angelatonio ED, et al. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 partcipants. J Am Coll Cardiol. 2010;55(19):2160–7.
Schwartz GG, Steg PG, Szarek M, Bittner VA, Diaz R. Peripheral artery disease and venous thromboembolic events after acute coronary syndrome, Role of Lipoprotein(a) and modification by alirocumab: prespecified analysis of the ODYSSEY OUTCOMES randomized clinical trial. Circulation. 2020;141:1608–17.
Patel AP, Wang M, Pirruccello P, et al. Lp(a) (Lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease. Atheroscler, Thromb Vasc Biol. 2021;41:465–74.
Jang AY, Han SH, Sohn S, Oh PC, Koh KK. Lipoprotein(a) and cardiovascular diseases. Revisited. Circ J. 2020;84:867–74.
Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2019;379(22):2097–107.
Fernandez-Prado R, Perez-Gomez MV, Ortiz A. Pelacarsen for lowering lipoprotein(a): implications for patients with chronic kidney disease. Clin Kidney J. 2020;13(5):753–7.
Tsimikas S, Karwatoska-Prokopczuk E, Gouni-Berthold I, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382(3):244–55.
Ruscica M, Greco MF, Ferri N, Corsini A. Gender difference in lipoprotein(a) concentration as a predictor of coronary revascularization in patients with known coronary artery disease. Eur Heart J. 2020;22(Suppl L):L53–6.
Goyal N, Narayanaswami P. Making sense of antisense oligionucleotides: a narrative review. Muscle Nerve. 2017;57(3):356–70.
Viney N, Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ. Antisense oligionucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. The Lancet. 2016;388:2239–53.
Tsimikas S, Viney NJ, Hughes SG. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. The Lancet. 2015;386:1472–83.
Tsimikas S. The re-emergence of lipoprotein(a0 in a broader clinical arean. Prog Cardiovasc Dis. 2016;59:135–44.
Novartis Pharmaceuticals. Assessing the impact of lipoprotein(a) lowering with TQJ230 on major cardiovascular events in patients with CVD (Lp(a)HORIZON). National Institutes of Health, US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04023552?term=tqj230&draw=2&rank=1. Accessed 28 Apr 2021.
Koren MJ, Moriarty PM, Neutel J, Baum SJ, Hernandez-Illas M. Abstract 13951: safety, tolerability and efficacy of single-dose AMG 890, a novel Sirna Targeting Lp(a), in healthy subjects and subjects with elevated Lp(a). Circulation. 2020;142(Suppl 3:A13951.
Stiekema LCA, Prange KHM, Hoogeveen RM, Verweij S, Kroon J. Potent lipoprotein(a) lowering following apolipoprotein(a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipoprotein(a). Eur Heart J. 2020;41(24):2262–71.
Burgess S, Ference BA, Staley JR, Freitag DF, Mason AM. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-Lowering therapies. A Mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619–617.
Madsen CM, Kamstrup PR, Langsted A, Varbo A, Nordestgaard BG. Lipoprotein(a)-lowering by 50 mg/dL (105 nmol/L) may be needed to reduce cardiovascular disease 20% in secondary prevention. Atherioscler Thromb Vasc Biol. 2020;40:255–66.
Benizri S, Gissot A, Martin A, Vialet B, Grinstaff MW. Bioconjugated oligionucelotides: recent developments and therapeutic applcations. Bioconjug Chem. 2019;30(2):366–83.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflicts of interest
Jennifer Hardy, Stephanie Niman, Rebecca F. Goldfaden, Majdi Ashchi, Mohannad Bisharat, Jessica Huston, Heather Hartmann, and Rushab Choksi declare they have no conflicts of interest that might be relevant to this manuscript.
Availability of data and material
Not applicable.
Ethics approval
Not applicable.
Code availability
Not applicable.
Consent to participate
Not applicable.
Consent to participate
Not applicable.
Author contributions
All authors contributed to the review. Rebecca Goldfaden had the idea for the article; Jennifer Hardy, Stephanie Niman, and Heather Hartmann performed the literature search and data analysis; and all authors drafted and/or critically revised the review. All authors read and approved the final manuscript.
Additional information
The original Online version of this article was revised: The Figure 2 was inadvertently published as Figure 2a and 2b.
Rights and permissions
About this article

Cite this article
Hardy, J., Niman, S., Goldfaden, R.F. et al. A Review of the Clinical Pharmacology of Pelacarsen: A Lipoprotein(a)-Lowering Agent. Am J Cardiovasc Drugs 22, 47–54 (2022). https://doi.org/10.1007/s40256-021-00499-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-021-00499-1