
Abstract
Decreasing low-density lipoprotein cholesterol (LDL-C) is one of the few established and proven principles for the prevention and treatment of atherosclerosis. The higher the individual cardiovascular risk, the higher the benefit of lipid-lowering pharmacotherapy. Therefore, treatment options are chosen based on a patient’s total cardiovascular risk. The latter depends not only on the levels of LDL-C but also on the presence of cardiovascular disease (CVD) and on the number and severity of other risk factors. Current guidelines recommend the lowering of LDL-C to 115 mg/dl (3 mmol/l) in patients with low and moderate risk. The LDL-C treatment target is <100 mg/dl (2.6 mmol/l) for patients at high risk and <70 mg/dl (1.8 mmol/l) for patients at very high risk. Although lifestyle measures remain a fundamental part of treatment, many patients require drug therapy to achieve their LDL-C targets. Statins are the drugs of choice, with other options including ezetimibe and the newly available monoclonal antibodies against PCSK9 (proprotein convertase subtilisin/kexin type 9). In some cases, bile acid-binding sequestrants and fibrates can also be considered. Nicotinic acid is no longer available in Germany. PCSK9 antibodies decrease LDL-C about 50–60 % and are well tolerated. Their effects on clinical endpoints are being investigated in large randomized trials. The aim of the present review is to summarize the current guidelines and treatment options for hypercholesterolemia. Moreover, we provide an appraisal of PCSK9 antibodies and propose their use in selected patient populations, particularly in those at very high cardiovascular risk whose LDL-C levels under maximally tolerated lipid-lowering therapy are significantly over their treatment target.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The concentration of low-density lipoprotein (LDL) in the plasma is causally linked to atherosclerosis and its clinical sequelae. |
The use of statins for LDL lowering has consistently been shown to reduce cardiovascular events and mortality. |
Inhibition of PCSK9 with monoclonal antibodies lowers LDL with unprecedented efficacy and the results of outcomes studies are expected next year. |
Before the results of outcome studies of PCSK9 inhibitors are available, we recommend their use in patients at extremely high risk of vascular events and with LDL cholesterol concentrations significantly above individual goals. |
1 Introduction
Although cardiovascular disease (CVD) remains the leading cause of death worldwide, in recent years, a clear decrease in cardiovascular mortality has been observed [1, 2]. Some of the reasons for this positive development are improvements in the acute treatment of cardiovascular events as well as improved control of cardiovascular risk factors, especially hypercholesterolemia. Regulatory agencies such as the US FDA as well as various guidelines consider low-density lipoprotein cholesterol (LDL-C) a causal risk factor for CVD [3–7]. This causal association between LDL-C and CVD is supported by (1) experimental findings on the pathogenesis of atherosclerosis [8]; (2) associations between monogenic [9–17] and polygenic [18–22] hypercholesterolemia levels and cardiovascular events; (3) epidemiologic studies [23–25]; and (4) interventional studies with cholesterol-lowering agents such as the HMG-CoA-reductase-inhibitors (statins) [26–30] and other agents [24, 31, 32].
2 Guidelines for Lipid-Lowering Therapy
The European Atherosclerosis Society/European Society of Cardiology (EAS/ESC) European and American Heart Association/American College of Cardiology (AHA/ACC) North American guidelines focus on LDL-C as the therapeutic target of clinical relevance [4, 6, 7]. Furthermore, they agree that the higher the risk of the patient, the more intensive the treatment should be. A recent consensus statement from the ACC and the National Lipid Association (NLA) has revised the previous “fire and forget” strategy that was focused on high-dose statin therapy. The new guidance recommends clinicians treat to individualized LDL-C targets, monitor LDL-C during therapy, use maximally tolerated doses of statins, and use combination therapy with ezetimibe as second-line and PCSK9 inhibitors as third-line therapy when LDL-C targets are not reached [33]. A decrease of LDL-C levels by about 1 mmol/l (40 mg/dl) is associated with a 22 % decrease in the relative risk for coronary events [30, 34]. This reduction occurs regardless of baseline risk. The higher the baseline risk of the patient, the higher the absolute benefits of treatment. If the risk is high, for example 25 % for a coronary event in the next 10 years, then a decrease in relative risk of 20 % translates to a decrease of the absolute risk of 5 %; that would mean 20 patients would have to be treated for 10 years to prevent one event. On the other hand, if the baseline risk is only 5 % in 10 years, the absolute risk reduction would be only 1 %, meaning that 100 patients would have to be treated for 10 years to prevent one event [35]. Moreover, the relative risk reduction achieved by a specific decrease in LDL-C levels is not the same in all age groups. It is higher in younger than in older individuals [24, 36]. Therefore, the lifetime risk and an early treatment of hypercholesterolemia are the focus of the current prevention strategies [37].
In Germany in 2009, in an effort to decrease lipid-lowering drug-related expense, it was decided that lipid-lowering treatment would be reimbursed only for individuals with a risk of experiencing a cardiovascular event in the next 10 years of ≥20 % [38]. Using this arbitrary cut-off, 90 % of the population (in which ~two-thirds of the myocardial infarctions will occur) is a priori excluded from treatment. A further substantial problem with this legislative approach is that it lacks the definition of a specific method to calculate risk.
Currently, the treatment targets proposed by the ESC and the EAS guidelines are recommended in Germany [4, 6]. They suggest four risk categories: low, moderate, high, and very high (Table 1). The LDL-C treatment targets are determined using the risk estimation system SCORE (Systematic Coronary Risk Evaluation) [39], the presence of known CVD or of markedly elevated single risk factors such as diabetes, severe hypertension, familial hypercholesterolemia, and moderate to severe chronic kidney disease (CKD). For all subjects, the LDL-C treatment target is 115 mg/dl (3.0 mmol/l), for those at high risk 100 mg/dl (2.6 mmol/l), and for those at very high risk <70 mg/dl (1.8 mmol/l) [4, 6].
3 Drug Therapy
3.1 Statins
Statins have consistently reduced cardiovascular events and are therefore, together with lifestyle modifications, the treatment of choice [4, 6, 7, 27, 30]. However, in some cases, statin monotherapy is not sufficient. One reason for this is that statins decrease LDL-C levels in a dose-dependent but not linear manner. The doubling of a statin dose is associated with an only ~6 % further decrease in LDL-C. Figure 1 shows the degrees of LDL-C reduction achieved with various statins [26, 40, 41]. In some patients with very high untreated LDL-C levels, LDL-C treatment targets cannot be achieved, even with the highest statin doses [42]. This is very commonly seen in patients with familial hypercholesterolemia [43, 44].

Statins are generally very well tolerated, and their good safety profile has been clearly documented over the years. However, they do have potential side effects such as abdominal discomfort; small, reversible increases in the transaminases; and muscle symptoms that, when present, may limit compliance with therapy [45–47]. Muscle complaints occur in 5–30 % of statin-treated patients. Predisposing factors are older age, infections, hypothyroidism, decreased renal function, liver disease, operations and muscle trauma, diabetes, alcohol consumption, pre-existing muscle disease, or use of specific concomitant medications [45–47]. Although the vast majority of patients with statin-associated muscle symptoms can eventually tolerate a statin at a low dose, many of these patients cannot reach their LDL-C treatment targets.
Moreover, statins increase the risk of developing type 2 diabetes by ~10 % [48]. However, the decrease in total and cardiovascular mortality associated with statin use clearly outweighs the disadvantage of potentially developing diabetes. Furthermore, the benefits of statin use in patients with diabetes have also been demonstrated. Therefore, statins are indicated in all patients with diabetes.
If the LDL-C treatment target is not achieved with the maximally tolerated statin dose, a combination therapy is indicated, with ezetimibe as first choice because the combination of simvastatin and ezetimibe has been shown to decrease the risk for myocardial infarction and stroke [32]. Other options include bile acid sequestrants and fibrates. Monoclonal antibodies against PCSK9 can also be used when the LDL-C levels remain significantly above the treatment target after two (some authors propose three) statins have been tried at their maximally tolerated dose.
3.2 Ezetimibe
Ezetimibe inhibits the Niemann-Pick C1 like 1 (NPC1L1) protein, a specific transporter of cholesterol, thus reducing cholesterol and plant sterol absorption [49]. The absorption of triglycerides and fat-soluble vitamins is not affected. Ezetimibe at a dose of 10 mg per day can decrease cholesterol absorption by 50 %. However, the decrease in circulating LDL-C levels is only ~20 % because of a compensatory increase in cholesterol synthesis in the liver [50]. Ezetimibe is mainly used in combination with statins. In SHARP (Study of Heart and Renal Protection), 9270 patients with CKD received either simvastatin 20 mg plus ezetimibe 10 mg daily or placebo. LDL-C was decreased by an average of 33 mg/dl (0.85 mmol/l) and the major atherosclerotic events by 17 % [51].
In the IMPROVE-IT trial, 18,141 patients with acute coronary syndrome and LDL-C <125 mg/dl (3.2 mmol/l) were treated with simvastatin 40 mg or simvastatin 40 mg plus ezetimibe 10 mg daily. The median average LDL-C levels in the group treated with simvastatin monotherapy was 70 mg/dl (1.8 mmol/l), and in the group treated with simvastatin plus ezetimibe it was 55 mg/dl (1.63 mmol/l). The event rate for the primary endpoint, a composite of cardiovascular death, myocardial infarction, unstable angina pectoris requiring hospitalization, coronary revascularization, or stroke, at 7 years was 32.7 % in the simvastatin–ezetimibe group, and 34.7 % in the simvastatin-monotherapy group. This translates to a relative risk reduction of 6.4 % (p = 0.016) in the intention-to-treat event rate of the composite primary endpoint and is consistent with the expected effect size. There was no decrease in cardiovascular or total mortality. The combination therapy potently reduced repeated events [52]. The study shows that a decrease in LDL-C is safe and beneficial irrespective of the mechanism through which the LDL-C is being lowered.
3.3 Monoclonal Antibodies against Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9)
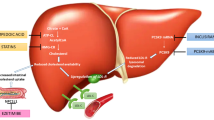
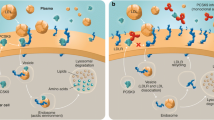
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is synthesized mainly in the liver but also in the kidneys, small intestine, and brain. In the plasma, PCSK9 circulates either bound to LDL or as free PCSK9 and regulates the catabolism of the LDL receptors [53]. Mutations of the PCSK9 gene, which increase its function (gain of function) cause an increased catabolism of the LDL receptors and are a cause, albeit rare, of familial hypercholesterolemia [53–55]. On the other hand, other PCSK9 gene variants cause decreased function of the gene. These are associated with lower LDL-C levels than those of the general population and incur a significantly decreased risk for the development of coronary heart disease [18]. When PCSK9 binds to the LDL receptor on the liver cell surface, the receptor can no longer dissociate itself from LDL-C in the endosome and both, LDL-C and LDL receptor, are catabolized in the lysosomes [53]. This decreases the amount of LDL receptors on the liver cell surface and subsequently increases LDL-C concentrations (Fig. 2). Statins increase the secretion of PCSK9, which in vivo may decrease the effects of statins [56–60]. Therefore, it seems reasonable to combine statins with PCSK9 antibodies. There is evidence that PCSK9 may also modulate other lipoprotein receptors [53], such as the very low-density lipoprotein (VLDL) receptor, the apolipoprotein E (ApoE)-receptor 2 (LRP8), and LRP1 [61–63]. Since lipoprotein(a) can be catabolized via these receptors [64], this is one of several potential explanations why PCSK9 antibodies, unlike statins, also decrease lipoprotein(a). Another proposed explanation for this effect would be that inhibition of PCSK9 suppresses the assembly of this lipoprotein. Further beneficial effects of PCSK9 inhibition independent from its effects on the lipoprotein receptors have been postulated, such as effects on inflammation and endothelial dysfunction [65].

Mechanism of action of PCSK9 antibodies (from Catapano and Papadopoulos [118]). a LDL binds to the LDL-Rs at the cell surface. The complexes of LDL and LDL-R are concentrated in specific areas of the cell membrane, the so-called coated pits. After their internalization, LDL and LDL-R dissociate in the endosome. The LDL-R then leaves the endosome and returns to the cell membrane before the former fuses with the lysosomes. There, the protein component of LDL is hydrolyzed and the LDL-C-associated cholesterol esters are catabolized with the help of the lysosomal acid lipase into fatty acids and cholesterol. The round-trip of the LDL-R to the endosome and back to the cell surface lasts about 12 min (of which, 3 min are spent in the cell and cell membrane, respectively and 6 min in the coated pits). Each LDL-R recirculates about 100 times in its life span. Free cholesterol can be used for the synthesis of cell membranes, steroid hormones, or—in the liver—for the synthesis of bile acids. It also suppresses the expression of LDL-Rs and HMG-CoA reductase and activates ACAT, an enzyme that esterifies free cholesterol for storage. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is mainly synthesized in the liver cells and regulates the degradation of the LDL-R. PCSK9 circulates in the blood either as free PCSK9 or bound on LDL. When PCSK9 binds to the LDL–LDL-R complex on the liver surface, a dissociation of the receptor is no longer possible, and they are both catabolized. This decreases the expression of the LDL-R and consequently increases the circulating LDL-C levels. b Monoclonal antibodies against PCSK9 bind circulating PCSK9, block the degradation of the LDL receptors, and thus increase the catabolism of LDL. LDL low-density lipoprotein, LDL-C low-density lipoprotein cholesterol, LDL-R LDL receptor
In 2015, two fully human monoclonal antibodies against PCSK9, alirocumab and evolocumab, were approved for use in patients. They bind circulating PCSK9 (Fig. 2) and decrease LDL-C concentrations by ~50 to 60 %, triglycerides by 1–18 %, and lipoprotein(a) by 23–31 % [66–68]. Other approaches to inhibit PCSK9 are in development, such as small interfering RNA (siRNA) [66–70], antisense oligonucleotides [71–73], and competitive inhibitors [63, 74].
Alirocumab is available in doses of 75 and 150 mg every 2 weeks, and evolocumab is available in doses of 140 mg every 2 weeks or 420 mg every 4 weeks (all administered subcutaneously) (Table 2). Both antibodies have been tested for efficacy and safety in a large number of studies (Table 3). Their effects are present either as monotherapy or co-administered with other lipid-lowering medications (statins up to their maximal dose or ezetimibe), and are not affected by diet modifications. Their efficacy has been tested and proven in various populations, such as those with statin intolerance [75, 76], familial hypercholesterolemia [77, 78], or those at high cardiovascular risk [79–81]. PCSK9 antibodies are well tolerated. The most common side effects are injection site reactions, nasopharyngitis, upper respiratory tract infections, nausea, and back pain, and—in some studies—also headache and fatigue. There was a statistically non-significant trend for more neurocognitive side effects in those using monoclonal antibodies against PCSK9 than in those receiving standard therapy [81, 82]. However, until now, no correlation between LDL-C levels under treatment and neurocognitive symptoms could be established. The question whether the use of PCSK9 antibodies is associated with cognitive side effects is specifically examined in the Ebbinghaus study with evolocumab [83].
In ODYSSEY Long Term, some effects of alirocumab on the levels of the fat-soluble vitamins E and K, which are almost exclusively transported in LDL, were observed [81]. More precisely, subjects in the group treated with alirocumab rather than those in the control group had circulating levels of vitamin E and K below the normal range; the concentrations of other fat-soluble vitamins and hormones remained unchanged. Likewise, in the DESCARTES study, absolute vitamin E levels decreased in the evolocumab-treated patients by 16 % after 52 weeks of treatment but actually increased by 19 % when normalized for cholesterol. No adverse effects were seen in steroid or gonadal hormones [84–86].
Long-term data on the safety of PCSK9 antibodies are currently sparse. However, it is reassuring that individuals who completely lack PCSK9 because of genetic mutations (loss-of function homozygotes) exhibit no pathologic phenotype, suggesting the long-term safety of very low PCSK9- and LDL-C serum concentrations [87, 88]. More information regarding the clinical efficacy and safety of PCSK9 antibodies will be provided by the long-term endpoint trials currently under way: FOURIER for evolocumab and ODYSSEY OUTCOMES for alirocumab. The results of the FOURIER trial are expected mid-2016. This study included approximately 27,000 patients who had experienced either a myocardial infarction or a stroke plus one major or at least two minor cardiovascular risk factors and LDL-C ≥70 mg/dl while receiving the maximally tolerated statin dose with or without ezetimibe. The primary endpoint is a composite of cardiovascular death, myocardial infarction, and hospitalization for unstable angina or coronary revascularization. ODYSSEY OUTCOMES included 18,000 patients with recent acute coronary syndrome. The primary endpoint is a composite of cardiovascular death, myocardial infarction, and hospitalization for unstable angina or stroke. Results are expected in 2018.
In the meantime, post hoc analyses of the phase III trials with evolocumab and alirocumab have been published and show promising findings regarding cardiovascular endpoints. In ODYSSEY Long Term, after 24 weeks of treatment, the cumulative incidence of cardiovascular events in the alirocumab group was ~50 % lower than in the group receiving standard therapy (1.7 vs. 3.3 %; hazard ratio 0.52, p = 0.02) [81]. Similar findings were shown in the post hoc analysis of the OSLER 1 and OSLER 2 studies with evolocumab. They also found a ~50 % decrease in the incidence of cardiovascular events (Fig. 3) [82]. Moreover, a recent meta-analysis of studies with PCSK9 antibodies showed a significant decrease, even in total mortality [66]. Altogether, while awaiting the results of the long-term endpoint trials, this preliminary evidence suggests one can be optimistic about the therapeutic potential of PCSK9 antibodies.

a Incidence of cardiovascular events in the OSLER studies (adapted from Sabatine et al. [82]). Cardiovascular events include death from cardiovascular causes, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, stroke, transient ischemic attack, and heart failure requiring hospitalization. b Incidence of cardiovascular events in the ODYSSEY Long Term study (adapted from Robinson et al. [81]). Death from coronary heart disease, nonfatal myocardial infarction, fatal or nonfatal ischemic stroke, or unstable angina requiring hospitalization
In Germany, two monoclonal antibodies are commercially available. They are both approved for a very wide spectrum of indications (Table 2). Although the effect of PCSK9 antibodies on clinical endpoints has not yet been proven, it is very likely—based on currently available data—that they may indeed decrease cardiovascular events. Given their high cost, we consider it prudent at this point to prescribe it only for patients at very high cardiovascular risk and after all other lipid-lowering agents (at least statins and ezetimibe) have been tried at their maximally tolerated dose. Moreover, the LDL-C level should be significantly and reproducibly higher than the treatment target. Since LDL-C levels may vary between measurements because of analytical and pre-analytical factors, the LDL-C values should be measured again at least once before PCSK9 therapy is initiated.
In patients with familial hypercholesterolemia [45] without cardiovascular events, the treatment target is an LDL-C value of <100 mg/dl. In our opinion, PCSK9 antibodies should be considered if the LDL-C values are ≥160 mg/dl under maximally tolerated conventional treatment.
Figure 4 and Table 4 show, based on results of the LURIC (Ludwigshafen Risk and Cardiovascular Health) study [89], that the risk for cardiovascular death in patients with CVD particularly depends on the number of their comorbidities.

Total mortality (a) and cardiovascular mortality (b) in 2272 clinically stable subjects in the Ludwigshafen Risk and Cardiovascular Health (LURIC) study without (n = 732) or with angiographically documented coronary heart disease (n = 1540) [89]. CVD was diagnosed if there was at least one lesion in a coronary artery that resulted in a ≥20 % reduction in lumen diameter. CVD + 0: no, CVD + 1 to CVD + 4: one to four of the following co-morbidities: familial hypercholesterolemia [90], previous myocardial infarction, diabetes mellitus [91], impaired renal function (calculated glomerular filtration rate <60 ml/min/1.72 m2) [92], moderate to severe heart failure (New York Heart Association classification III and IV). CVD cardiovascular disease
Therefore, in “secondary prevention” (patients with CVD, particularly with unequivocally diagnosed coronary heart disease), we propose treatment with PCSK9 antibodies if the LDL-C, under well-documented, maximally tolerated conventional lipid-lowering pharmacotherapy, remains ≥130 mg/dl and at least two of the following factors are present:
-
familial hypercholesterolemia [90].
-
previous myocardial infarction, clinically evident progression of coronary heart disease, or atherosclerosis of other vessels.
-
diabetes mellitus [91].
-
moderate to severe CKD (glomerular filtration rate [GFR] <60 ml/min/1.73 m2) [92].
-
heart failure (mainly ischemic) New York Heart Association (NYHA) classification III–IV.
This preliminary stratification is easy to use in clinical practice and can of course be modified in individual cases within the frame set by the approved indications for PCSK9 antibodies. For instance, patients in whom coronary heart disease progresses exceptionally quickly (e.g. repeated myocardial infarctions) and who do not reach LDL-C goals despite other lipid-lowering medicines may qualify for instant initiation of treatment. If the criteria proposed above are used, ~1 % (shaded fields in Table 5) of the patients with coronary heart disease would be considered as candidates for treatment with PCSK9 antibodies (on top of the maximally tolerated lipid-lowering therapy). Assuming a prevalence of 6 million patients with coronary heart disease in Germany [93], this would include 60,000 patients, which is approximately the patient population with an indication for LDL apheresis according to the guidance of the Federal Joint Committee.
Currently, the annual cost of PCSK9 antibody treatment in Germany is around €8500 per patient. Since even treating 60,000 patients would place a significant cost burden on the healthcare system, we sought to obtain a rough estimate of the cost effectiveness of treatment in terms of life-years saved. In this analysis, we assumed that patients with coronary heart and two comorbidities were treated either with conventional, maximal lipid-lowering therapy or, in addition, with PCSK9 antibodies. Cardiovascular mortality at 10 years was set at approximately 35 % in the conventional treatment group and 50 % lower with a PCKS9 antibody, with no effect on non-cardiovascular causes of death. We also accounted for cost savings from the prevention of non-fatal coronary and cerebrovascular events using a ratio of three non-fatal events per one fatal event and costs of €13,000 and €47,000 per non-fatal myocardial infarction or non-fatal cerebrovascular event, respectively. This provisional calculation resulted in costs per life-year saved of ~€100,000. We wish to emphasize that an in-depth health economic analysis is beyond the scope of this review article and will truly be appropriate only after the results of the ongoing outcome trials with PCSK9 antibodies have become available. In the meantime, it should be considered that the expenses for LDL apheresis, and consequentially the costs per life-years saved, are four to five times higher than for PCSK9 antibody therapy.
For both medical reasons and based on the phrasing of the Policy Methods of Contractual Medical Care (Richtlinie Methoden vertragsärztliche Versorgung) of the Federal Joint Committee (Gemeinsamer Bundesausschuss) of 16 May 2015 (http://www.g-ba.de/downloads/62-492-1022/MVV-RL_2015-02-19_iK-2016-05-16.pdf), we therefore suggest that LDL apheresis should be considered only if LDL-C cannot adequately be lowered within a period of at least 12-months during which diet, oral lipid-lowering therapy, and PCSK9 antibodies have been tried. A rather justified exception applies for patients with homozygous familial hypercholesterolemia. In these patients, LDL apheresis can be initiated before other treatments have been tried [94].
3.4 Other Lipid-Lowering Agents
Nicotinic acid is no longer available in Germany, after the studies AIM-HIGH [95] and HPS2-THRIVE (Heart Protection Study 2- Treatment of HDL [high-density lipoprotein] to Reduce the Incidence of Vascular Events) [96] showed that adding nicotinic acid to statins does not bring any significant clinical benefit to patients. Moreover, in the HPS2-THRIVE study, the group receiving nicotinic acid experienced severe side effects more often, including intracranial and gastrointestinal bleeding, myopathies, infections, and diabetes.
The orally administered bile acid sequestrants are therapeutic options for the treatment of severe hypercholesterolemia or statin intolerance. However, they are often not well tolerated because of gastrointestinal side effects. Bile acid sequestrants were evaluated in LRC-CPPT (Lipid Research Clinics Coronary Primary Prevention Trial) and were shown to reduce CVD events in the patients studied [97]. Whether the addition of bile acid sequestrants to statins reduces clinical endpoints remains unclear. Fibrates can be used as monotherapy for the treatment of hypertriglyceridemia or in combination with other lipid-lowering agents in patients with mixed dyslipidemia. The effects of fibrates on cardiovascular endpoints is disputed, but recent studies examining a potential benefit when added to statins had negative results [98]. Mipomersen is an antisense oligonucleotide against Apo-B that is administered subcutaneously and decreases LDL-C by up to 30 % [99]. It is approved in the USA but not in Europe for the treatment of homozygous hypercholesterolemia. Lomitapide is an oral inhibitor of the microsomal triglyceride transfer protein (MTP) and decreases LDL-C by ≥40 % [100]. The most important side effect of lomitapide is a considerable increase in liver fat (hepatic steatosis). It is approved only for the treatment of homozygous hypercholesterolemia both in Europe (since 2013) and in the USA (since 2012). Its effect on cardiovascular endpoints is unknown. The cholesterylester transfer protein (CETP) inhibitors were assumed to decrease cardiovascular events since they robustly increase HDL-cholesterol (HDL-C) levels. However, this hope has not been realized until now. The development of torcetrapib was stopped in 2006 after the phase III trial ILLUMINATE (Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events) showed that it significantly increased both total and cardiovascular mortality [101]. The endpoint trial Dal-OUTCOMES (dalcetrapib 600 mg vs. placebo in patients with acute coronary syndrome) was stopped prematurely for futility in May 2012 [102]. An endpoint trial with evacetrapib [103] was also stopped prematurely in October 2015 [104]. The REVEAL (Randomized Evaluation of the Effects Anacetrapib through Lipid modification) study with anacetrapib is ongoing [105].
4 Conclusions
Regarding LDL-C, there is compelling evidence for the ‘the lower—the better’ thesis. Statins in addition to dietary and lifestyle changes remain the primary therapeutic option to reduce LDL-C. There is now also a solid scientific basis for initiation of treatment with ezetimibe in patients who do not reach LDL-C goals with the maximally tolerated dose of a potent statin. Although endpoint studies are not yet completed, there is mounting evidence that PCSK9 antibodies markedly reduce not only LDL-C but also cardiovascular risk. However, even in the absence of the results of the long-term endpoint trials, the initiation of PCSK9 antibodies is justified and sufficiently founded in patients at very high cardiovascular risk whose serum LDL-C levels are very high despite optimal oral lipid-lowering therapy.
References
Ford ES, Ajani UA, Croft JB, et al. Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N Engl J Med. 2007;356:2388–98.
Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366:54–63.
Mitka M. Amid lingering questions. FDA reprieves LDL cholesterol-lowering medication. JAMA. 2009;301:813–5.
Perk J, De Backer G, European Gohlke H, et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J. 2012;33:1635–701.
Rasnake CM, Trumbo PR, Heinonen TM. Surrogate endpoints and emerging surrogate endpoints for risk reduction of cardiovascular disease. Nutr Rev. 2008;66:76–81.
Reiner Z, Catapano AL, De Backer G, et al. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32:1769–818.
Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;2014:S46–8.
Tsimikas S, Miller YI. Oxidative modification of lipoproteins: mechanisms, role in inflammation and potential clinical applications in cardiovascular disease. Curr Pharm Des. 2011;17:27–37.
Brown MS, Goldstein JL. A receptor mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47.
Marks D, Thorogood M, Neil HA, et al. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. 2003;168:1–14.
Kolansky DM, Cuchel M, Clark BJ, et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol. 2008;102:1438–43.
Widhalm K, Binder CB, Kreissl A, et al. Sudden death in a 4-year-old boy: a near-complete occlusion of the coronary artery caused by an aggressive low-density lipoprotein receptor mutation (W556R) in homozygous familial hypercholesterolemia. J Pediatr. 2011;158:167.
Raal FJ, Pilcher GJ, Panz VR, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124:2202–7.
Macchiaiolo M, Gagliardi MG, Toscano A, et al. Homozygous familial hypercholesterolaemia. Lancet. 2012;379:1330.
Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–90.
Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–57.
Klose G, Laufs U, Marz W, et al. Familial hypercholesterolemia: developments in diagnosis and treatment. Dtsch Arztebl Int. 2014;111:523–9.
Cohen JC, Boerwinkle E, Mosley TH, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60:2631–9.
Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80.
Stitziel NO, Won HH, Morrison AC, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371:2072–82.
Ference BA, Majeed F, Penumetcha R, et al. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial Mendelian randomization study. J Am Coll Cardiol. 2015;65:1552–61.
Kannel WB, Castelli WP, Gordon T, et al. Serum cholesterol, lipoproteins, and the risk of coronary heart disease. The Framingham study. Ann Intern Med. 1971;74:1–12.
Law MR, Wald NJ, Thompson SG. By how much and how quickly does reduction in serum cholesterol concentration lower risk of ischemic heart disease. BMJ. 1994;308:367–73.
Lewington S, Whitlock G, Clarke R, et al. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–39.
Law MR, Wald NJ, Rudnicka AR. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta-analysis. BMJ. 2003;326:1423.
Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78.
Genser B, März W. Low density lipoprotein cholesterol, statins and cardiovascular events: a meta-analysis. Clin Res Cardiol. 2006;95:393–404.
Boekholdt SM, Hovingh GK, Mora S, et al. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta-analysis of statin trials. J Am Coll Cardiol. 2014;64:485–94.
Fulcher J, O’Connell R, Voysey M, et al. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385:1397–405.
Buchwald H, Varco RL, Matts JP, et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia: report of the Program on the Surgical Control of Hyperlipidemias. N Engl J Med. 1990;323:946–55.
Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97.
Lloyd-Jones DM, Morris PB, Ballantyne CM, et al. 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology Task Force on clinical expert consensus documents. J Am Coll Cardiol. 2016. doi:10.1016/j.jacc.2016.03.519.
Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81.
Laufs U, Weintraub WS, Packard CJ. Beyond statins: what to expect from add-on lipid regulating therapy? Eur Heart J. 2013;34:2660–5.
Cholesterol Treatment Trialists C, Mihaylova B, Emberson J, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–90.
Packard CJ, Weintraub WS, Laufs U. New metrics needed to visualize the long-term impact of early LDL-C lowering on the cardiovascular disease trajectory. Vascul Pharmacol. 2015;71:37–9.
Kassenärztliche Bundesvereinigung. Schnellübersicht der Kassenärztlichen Bundesvereinigung und des GKV-Spitzenverbandes zur Verordnungsfähigkeit von Arzneimitteln nach der Arzneimittel-Richtlinie (AM-RL), § 92 Abs.1 Satz 2 Nr. 6 SGB V, gültig ab 01.04.2009 (Stand: 19.03.2015). http://www.kbv.de/media/sp/Schnelluebersicht_Verordnungsfaehigkeit_Arzneimittel.pdf. 2009.
European Society of Cardiology. ESC-HeartScore. Available online from http://www.heartscore.org. Accessed 19 Feb 2016.
Mukhtar RY, Reid J, Reckless JP. Pitavastatin. Int J Clin Pract. 2005;59:239–52.
Weng TC, Yang YH, Lin SJ, et al. A systematic review and meta-analysis on the therapeutic equivalence of statins. J Clin Pharm Ther. 2010;35:139–51.
Jones PH, Nair R, Thakker KM. Prevalence of dyslipidemia and lipid goal attainment in statin-treated subjects from 3 data sources: a retrospective analysis. J Am Heart Assoc. 2012;1:e001800.
Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The Netherlands. Atherosclerosis. 2010;209:189–94.
Stein EA, Strutt K, Southworth H, et al. Comparison of rosuvastatin versus atorvastatin in patients with heterozygous familial hypercholesterolemia. Am J Cardiol. 2003;92:1287–93.
Laufs U, Scharnagl H, Halle M, et al. Treatment options for statin-associated muscle symptoms. Dtsch Arztebl Int. 2015;112:748–55.
Laufs U, Scharnagl H, März W. Statin intolerance. Curr Opin Lipidol. 2015;26:492–501.
Stroes ES, Thompson PD, Corsini A, et al. Statin-associated muscle symptoms: impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J. 2015;36:1012–22.
Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–42.
Lyseng-Williamson KA. Ezetimibe/simvastatin: a guide to its clinical use in hypercholesterolemia. Am J Cardiovasc Drugs. 2012;12:49–56.
Sudhop T, Lutjohann D, Kodal A, et al. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation. 2002;106:1943–8.
Baigent C, Landray MJ, Reith C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet. 2011;377:2181–92.
Murphy SA, Cannon CP, Blazing MA, et al. Reduction in total cardiovascular events with ezetimibe/simvastatin post-acute coronary syndrome: the IMPROVE-IT trial. J Am Coll Cardiol. 2016;67:353–61.
Seidah NG, Awan Z, Chretien M, et al. PCSK9: a key modulator of cardiovascular health. Circ Res. 2014;114:1022–36.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6.
Varret M, Rabes JP, Saint-Jore B, et al. A third major locus for autosomal dominant hypercholesterolemia maps to 1p34.1-p32. Am J Hum Genet. 1999;64:1378–87.
Careskey HE, Davis RA, Alborn WE, et al. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res. 2008;49:394–8.
Costet P, Hoffmann MM, Cariou B, et al. Plasma PCSK9 is increased by fenofibrate and atorvastatin in a non-additive fashion in diabetic patients. Atherosclerosis. 2010;212:246–51.
Dong B, Wu M, Li H, et al. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. 2010;51:1486–95.
Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–9.
Welder G, Zineh I, Pacanowski MA, et al. High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res. 2010;51:2714–21.
Canuel M, Sun X, Asselin MC, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One. 2013;8:e64145.
Poirier S, Mayer G, Benjannet S, et al. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem. 2008;283:2363–72.
Shan L, Pang L, Zhang R, et al. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Commun. 2008;375:69–73.
März W, Beckmann A, Scharnagl H, et al. Heterogenous lipoprotein (a) size isoforms differ by their interaction with the low density lipoprotein receptor and the low density lipoprotein receptor-related protein/a2-macroglobulin. FEBS Lett. 1993;325:271–5.
Urban D, Poss J, Bohm M, et al. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol. 2013;62:1401–8.
Lipinski MJ, Benedetto U, Escarcega RO, et al. The impact of proprotein convertase subtilisin-kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta-analysis. Eur Heart J. 2015. doi:10.1093/eurheartj/ehv563 [Epub ahead of print].
Navarese EP, Kolodziejczak M, Schulze V, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta-analysis. Ann Intern Med. 2015;163:40–51.
Shimada YJ, Cannon CP. PCSK9 (Proprotein convertase subtilisin/kexin type 9) inhibitors: past, present, and the future. Eur Heart J. 2015;36:2415–24.
Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60–8.
Frank-Kamenetsky M, Grefhorst A, Anderson NN, et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci. 2008;105:11915–20.
Graham MJ, Lemonidis KM, Whipple CP, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007;48:763–7.
Gupta N, Fisker N, Asselin MC, et al. A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo. PLoS One. 2010;5:e10682.
Lindholm MW, Elmen J, Fisker N, et al. PCSK9 LNA antisense oligonucleotides induce sustained reduction of LDL cholesterol in nonhuman primates. Mol Ther. 2012;20:376–81.
Zhang Y, Eigenbrot C, Zhou L, et al. Identification of a small peptide that inhibits PCSK9 protein binding to the low density lipoprotein receptor. J Biol Chem. 2014;289:942–55.
Stroes E, Colquhoun D, Sullivan D, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541–8.
Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA. 2012;308:2497–506.
Raal FJ, Honarpour N, Blom DJ, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:341–50.
Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:331–40.
Cannon CP, Cariou B, Blom D, et al. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J. 2015;36:1186–94.
Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: The ODYSSEY COMBO I study. Am Heart J. 2015;169(906–915):e913.
Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99.
Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–9.
Anon. Evaluating PCSK9 Binding antiBody Influence oN coGnitive HeAlth in High cardiovascUlar Risk Subjects (EBBINGHAUS) [NCT02207634]. Available online from https://clinicaltrials.gov. Accessed 19 Feb 2016.
Blom DJ, Djedjos CS, Monsalvo ML, et al. Effects of evolocumab on vitamin e and steroid hormone levels: results from the 52-week, phase 3, double-blind, randomized placebo-controlled DESCARTES study. Circ Res. 2015;117:731–41.
Colhoun HM, Ginsberg HN, Robinson JG, et al. Alirocumab effect on glycemic measures in individuals without diabetes at baseline. Circulation. 2015;132:A16863.
Sattar N, Preiss D, Blom D, et al. Evaluation of the one-year efficacy, safety and glycaemic effects of evolocumab (AMG 145) in 4,802 subjects with, at high risk for, or at low risk for, diabetes mellitus. Diabetologia. 2015;58:S79.
Hooper AJ, Marais AD, Tanyanyiwa DM, et al. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193:445–8.
Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79:514–23.
Winkelmann BR, März W, Boehm BO, et al. Rationale and design of the LURIC study–a resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics. 2001;2:S1–73.
Defesche JC, Lansberg PJ, Umans-Eckenhausen MA, et al. Advanced method for the identification of patients with inherited hypercholesterolemia. Semin Vasc Med. 2004;4:59–65.
American Disábetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2010;33(Suppl 1):S62–9.
Shlipak MG, Matsushita K, Arnlov J, et al. Cystatin C versus creatinine in determining risk based on kidney function. N Engl J Med. 2013;369:932–43.
Gosswald A, Schienkiewitz A, Nowossadeck E, et al. Prevalence of myocardial infarction and coronary heart disease in adults aged 40–79 years in Germany: results of the German Health Interview and Examination Survey for Adults (DEGS1). Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2013;56:650–5.
Bundesausschuss G. Richtlinie des Gemeinsamen Bundesausschusses zu Untersuchungs- und Behandlungsmethoden der vertragsärztlichen Versorgung. Richtlinie Methoden vertragsärztliche Versorgung, in der Fassung vom 17. Januar 2006, veröffentlicht im Bundesanzeiger 2006 Nr. 48 (S. 1 523), in Kraft getreten am 1. April 2006, zuletzt geändert am 19. Februar 2015, veröffentlicht im Bundesanzeiger (BAnz AT 15.05.2015 B7), in Kraft getreten am 16. Mai 2015.
Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34:1279–91.
The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA. 1984;251:351–64.
Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–61.
Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006.
Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6.
Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–99.
Nicholls SJ, Brewer HB, Kastelein JJ, et al. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA. 2011;306:2099–109.
Eli Lilly. Lilly to Discontinue Development of Evacetrapib for High-Risk Atherosclerotic Cardiovascular Disease Oct 12, 2015. Available online from https://investor.lilly.com/releasedetail.cfm?ReleaseID=936130. Accessed 19 Feb 2016.
Anon. Randomized EValuation of the Effects of Anacetrapib Through Lipid-modification (REVEAL) [NCT01252953]. Available online from https://clinicaltrials.gov. Accessed 19 Feb 2016.
Roth EM, McKenney JM. ODYSSEY MONO: effect of alirocumab 75 mg subcutaneously every 2 weeks as monotherapy versus ezetimibe over 24 weeks. Future Cardiol. 2015;11:27–37.
Koren MJ, Lundqvist P, Bolognese M, et al. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2531–40.
Moriarty PM, Thompson PD, Cannon CP, et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9:758–69.
Bays H, Gaudet D, Weiss R, et al. Alirocumab as add-on to atorvastatin versus other lipid treatment strategies: ODYSSEY OPTIONS I randomized trial. J Clin Endocrinol Metab. 2015;100:3140–8.
Farnier M, Jones P, Severance R, et al. Efficacy and safety of adding alirocumab to rosuvastatin versus adding ezetimibe or doubling the rosuvastatin dose in high cardiovascular-risk patients: the ODYSSEY OPTIONS II randomized trial. Atherosclerosis. 2016;244:138–46.
Robinson JG, Nedergaard BS, Rogers WJ, et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014;311:1870–82.
Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36:2996–3003.
Ginsberg HN, Rader DJ, Raal FJ, et al. ODYSSEY HIGH FH: efficacy and safety of alirocumab in patients with severe heterozygous familial hypercholesterolemia. Circulation. 2014;130:2119.
Bruckert E, Blaha V, Stein EA, et al. Trial assessing long-term use of PCSK9 inhibition in patients with genetic LDL disorders (TAUSSIG): efficacy and safety in patients with homozygous familial hypercholesterolemia receiving lipid apheresis. Circulation. 2014;130:A17016.
Blom DJ, Hala T, Bolognese M, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809–19.
Schwartz GG, Bessac L, Berdan LG, et al. Effect of alirocumab, a monoclonal antibody to PCSK9, on long-term cardiovascular outcomes following acute coronary syndromes: rationale and design of the ODYSSEY outcomes trial. Am Heart J. 2014;168:682–9.
Walma EP, Wiersma TJ. NHG-Standpunt Diagnostiek en behandeling van familiaire hypercholesterolemie. Huisarts Wet. 2006;49:202–4.
Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis. 2013;228:18–28.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Winfried März is employed with Synlab Holding Deutschland GmbH, has received research grants from Aegerion Pharmaceuticals, AMGEN, Astrazeneca, Danone Research, Sanofi/Genzyme, Pfizer, and BASF, and has received speaker honoraria from Aegerion Pharmaceuticals, AMGEN, Astrazeneca, Danone Research, Sanofi/Genzyme, Pfizer, BASF, Hoffmann LaRoche, MSD, and Sanofi. Ioanna Gouni-Berthold has received consulting and speaker fees and support for educational activities from Amgen, Sanofi and Genzyme. Günther Silbernagel has received a research grant, consulting fees, and support for travel from Amgen. Ulf Landmesser has received speaker fees from MSD, Pfizer, Amgen, Sanofi, Roche, and Berlin-Chemie. Hans Dieplinger has received speaker honoraria and consulting fees from Amgen and Sanofi-Aventis. Eberhard Windler has received honoraria for consulting and lectures from AMGEN, AstraZenca, MSD, Pfizer, Sanofi, and Unilever. Ulrich Laufs has received speaker honoraria and consulting fees from Amgen, MSD, Sanofi, and Pfizer. Hubert Scharnagl, Alexander Dressel, and Tanja B. Grammer have no conflicts of interest that might be relevant to the contents of this manuscript.
Rights and permissions
About this article

Cite this article
März, W., Scharnagl, H., Gouni-Berthold, I. et al. LDL-Cholesterol: Standards of Treatment 2016: A German Perspective. Am J Cardiovasc Drugs 16, 323–336 (2016). https://doi.org/10.1007/s40256-016-0179-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-016-0179-y