
Abstract
Background
Regarding the role of gut microbial dysbiosis in hyperglycemia, we aimed to compare the main gut bacterial composition among type 1 and type 2 diabetic patients and healthy non-diabetic adults.
Methods
A total of 110 adult subjects (49 patients diagnosed with type 2 diabetes, 21 patients diagnosed with type 1 diabetes and 40 healthy persons) were included in this case-control study. The intestinal microbiota composition was investigated by quantitative real-time polymerase chain reaction (qPCR) method targeting bacterial 16S rRNA gene. Comparison between three groups was done using one-way analysis of variance.
Results
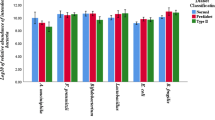
The participants’ mean age in the type 1 diabetes, type 2 diabetes and control groups was 35.4, 57.2 and 38.0 years, respectively. Higher level of Escherichia, Prevotella and Lactobacillus was observed in both type 1 and type 2 diabetic patients compared with the healthy group (P ˂0.001). In contrast, bacterial load of Bifidobacterium, Roseburia and Bacteroides was higher in healthy control group (P < 0.05). Faecalibacterium was significantly lower in type 1 diabetic patients compared with the other two groups (P ˂0.001). No significant difference was found in Akkermansia level among three groups.
Conclusions
Gut microbial alterations have been observed among patients suffering from type 1 and type 2 diabetes mellitus and healthy control adults. Butyrate producing genera including Roseburia and Faecalibacterium decreased while Escherichia, Prevotella and Lactobacillus increased in diabetic patients compared to healthy subjects. Modulating approaches of gut microbiota composition could be helpful in diabetes management.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Diabetes mellitus is one of the most prevalent metabolic disorders worldwide, which recognized with hyperglycemia and glucose intolerance. Diabetes usually divided into two groups of type 1 and type 2 diabetes. Genetic and environmental factors, like a sedentary lifestyle and high energy diet, exposed people to the risk of diabetes [1]. Recent studies proposed gut microbiota as an effective factor in diabetes pathogenesis [2, 3]. Gut microbiota, the wide range of symbiotic microorganisms that live in the intestine, makes a dynamic ecosystem inside the host body. The composition and function of the gut microbiota depends on the host genome, dietary intake, and other lifestyle factors [4,5,6].
Several host metabolic pathways, including interactive host-microbiota metabolic endocannabinoids, production of bile acids, short chain fatty acids (SCFAs), biogenic amines and xenometabolites are affected by the host-microbiota interactions [4]. Based on the several investigations, enteroendocrine cell metabolism and the endocannabinoid system are controlled by the gut microbiota. Moreover, gut microbiota constitutes a link among gut barrier dysfunction, metabolic endotoxemia and low-grade inflammation [7]. Identification of these links can lead to new therapeutic strategies to prevent diseases and improve health [4, 8,9,10].
Low-grade inflammation has been suggested as a potential contributing factor for diabetes. The gut microbiota is a rich source of the components such as lipopolysaccharides (LPS) that in interaction with the immune cells of the host can bind to immune receptors and induce pro-inflammatory response. Thus, gut microbiota-derived LPS and peptidoglycans are factors which involved in the onset and progression of inflammation in peripheral tissues, metabolic endotoxemia and metabolic diseases [11, 12].
In recent years, higher rates of type 1 diabetes have been reported, that are not explained by genetic cause, but it could assign to changes in epigenetic and environmental factors such as lifestyle, diet, hygiene, and antibiotic usage that can induce alteration in microbiota composition [13]. Prevention of mucin synthesis by increase in the abundance of non-butyrate-producing lactate-utilizing bacteria may lead to the β-cell autoimmunity and in consequence type 1 diabetes [14]. Moreover, involvement of microbiota in critical regulating pathways such as insulin signaling and glucose homeostasis, contributes to the link between type 2 diabetes and gut microbiome [11, 15]. Gut microbial dysbiosis, decrease in butyrate-producing bacteria and increase in opportunistic pathogens, has been reported in diabetic patients [11]. Regarding the necessity of better understanding the gut microbiota alteration in diabetic patients for glycemic control, we aimed to compare the main gut bacterial composition in type 1 and type 2 diabetic patients and healthy non-diabetic adults.
Materials and methods
In this case-control study, 110 subjects were recruited from March 2017 to December 2017. Subjects were recruited via advertising at the endocrine clinics. 49 type 2 diabetics, 21 type 1 diabetics and 40 healthy non-diabetic subjects aged between 20 and 65 years entered this study. To ensure comparable data, we considered exclusion criteria included pregnancy and lactation, smoking, use of corticosteroids, antibiotics, prebiotics or probiotics during the 3 month prior to the study and suffering from co-morbidities including cardiovascular disease, kidney and liver disorders, gastrointestinal disorders, inflammatory bowel diseases and cancer and history of acute and chronic diarrhea over the last month. The written informed consent was obtained from all participants. All procedures were approved by the ethical committee of National Institute for Medical Research Development (NIMAD).
After collecting demographic information, anthropometric measurements were done. Participant’s weight was measured with an accuracy of 0.1 kg with light clothes using a digital scale (Seca, Germany) and height was measured with a precision of 0.5 cm without shoes using a stadiometer. Then body mass index (BMI) has been calculated as body weight in kilograms divided by the square of height in meter (kg/m2).
Blood samples were taken after 12–14 h of overnight fasting from participates. The blood serum was separated by centrifuging at 1300 g for 10 min at the room temperature immediately and stored at −80 °C freezer until analysis. Serum fasting blood sugar (FBS), total cholesterol (TC), triglyceride (TG), high-density lipoprotein cholesterol (HDL), low-density lipoprotein cholesterol (LDL), very low density lipoprotein cholesterol (VLDL) and high-sensitivity C-reactive protein (hsCRP) concentrations were measured by Roche kits using auto-analyzer instrument (Hitachi, Cobas C 311, Roche Diagnostics GmbH). Serum insulin concentration was measured by an enzyme immunoassay kit (Monobind Inc., Lake Forest, CA, USA).
Fresh faecal sample have been collected in sterile cups, stored immediately in ice packs and brought to the laboratory within 2 h. Samples were stored at −80 °C for faecal microbial quantification. Extraction of total bacterial DNA from 200 mg of each faecal sample has been done using QIAamp DNA Stool Mini kit (QIAGEN, GmbH, Germany) according to the manufacturer’s protocol. The extracted DNA purity and concentration were determined by Nanodrop spectrophotometer (Thermo Scientific NanoDrop, USA). All the extracted DNAs were stored at −20 °C until further analysis.
Genus-specific sequences of primers targeted the bacterial 16S rRNA genes used in the current study. Specificity of the primers was evaluated in silico using the nucleotide BLAST in NCBI. The specific primers sequences are shown in Table 1 [16,17,18,19,20,21,22,23].
Quantitative real-time PCR was performed in triplicate using Roche LightCycler® 96 System (Switzerland) and Sybr green Master Mix (Takara Bio, Shiga, Japan). The used thermal cycling conditions were as follows: an initial DNA denaturation step at 95 °C for 1 min; 40 cycles of denaturation at 95 °C for 5 s; primer annealing at 55 °C for 30 s; extension at 72 °C for 30 s. Finally, melting curve analysis was performed by slowly cooling the PCRs from 95 °C to 60 °C for confirming the specificity of the amplification products. The bacterial concentration in each sample was calculated by comparing the obtained threshold cycle values with the standard curves constructed for each experiment using serial 10-fold dilutions of bacterial genomic DNA of standard Ecoli (ATCC 25922) with known concentration. After that, bacterial copy numbers of the eight bacterial genera in 1 g of faeces were determined.
Statistical analysis was performed by IBM SPSS version 22.0 (SPSS Inc. Chicago, IL, USA). A P value <0.05 was considered as the level of significance. Data were expressed as mean ± standard deviation or median ± inter quartile range. The normality distribution for different variables was tested by the Kolmogorov–Smirnov test. For hs-CRP which did not follow normal distribution, log transformation was done. Chi-square test was used to test the equality in distribution of males and females among three groups. Comparison between three groups was done using one-way ANOVA or Kruskal-Wallis Test for variables with normal and non-normal distribution, respectively. Following one-way ANOVA, Bonferroni post hoc test was run for pairwise multiple comparisons.
Results
The participants’ mean age in the type 1 diabetes, type 2 diabetes and control groups was 35.4, 57.2 and 38.0 years, respectively. The mean age of adult patients with type 2 diabetes was significantly higher compared with the type 1 diabetics and healthy controls (P ˂0.001). The three groups were comparable with regard to sex distribution (P > 0.05). Use of diabetes medications, including metformin and insulin, were observed in 14% and 95% of participants with type 1 diabetes, also in 67% and 30% of type 2 diabetic patients, respectively. Moreover, 14% and 28% of type 1 and type 2 diabetic patients reported the use of lipid-lowering statins. Weight and BMI were significantly different among groups, patients with type 1 diabetes had lower weight compared with the two other groups (P < 0.05). The anthropometric indices and biochemical variables of the non-diabetic adults and those with type 1 and 2 diabetes are presented in Table 2. The diabetic groups had elevated concentration of FBS compared to the healthy group (P ˂0.001). However, serum insulin concentration was not significantly different among the groups (P > 0.05). TC and LDL-C were significantly higher in non-diabetic group compared with the other groups (P < 0.05), while HDL-C was also significantly higher in the control group (P ˂0.001). No statistically significant differences in TG and VLDL were observed among the study groups (P > 0.05). Hs-CRP concentration was significantly lower in non-diabetic participants compared to the diabetic patients (P ˂0.001).
Within Proteobacteria phylum, bacterial load of Escherichia in fecal samples of non-diabetic subjects was significantly lower compared to the diabetic patients (P ˂0.001). Within Bacteroidetes phylum, the bacterial load of Prevotella was significantly higher whereas the quantity of Bacteroides was significantly lower in type 1 and type 2 diabetic patients compared to the non-diabetic group (P ˂0.001).
Roseburia, a butyrate-producing genus, was significantly increased in non-diabetic subjects (P = 0.02). Faecalibacterium, another butyrate-producing genus within Firmicutes phylum, was significantly lower in type 1 diabetic patients compared with the other two groups (P ˂0.001). In diabetic patients, the bacterial number of Lactobacillus was significantly enriched compared with the healthy control group (P ˂0.001). No significant difference was found in Akkermansia level among three groups (P > 0.05). Within Actinobacteria, the number of Bifidobacterium was significantly higher in non-diabetic subjects (P = 0.04) (Table 3).
Discussion
In this case control study, bacterial load determination in faeces showed higher level of Escherichia, Prevotella and Lactobacillus in both type 1 and type 2 diabetic patients compared with the healthy group. In contrast, bacterial load of Bifidobacterium, Akkermansia, Roseburia, Faecalibacterium and Bacteroides was higher in healthy control group as expected based on previous investigations.
Several studies to date have investigated the gut microbiota composition in diabetic patients, as the most comprehensive ones we could mention to Qin et al. and Karlsson et al. studies, both of them indicated lower abundance of butyrate-producing bacteria including Roseburia and Faecalibacterium in diabetic patients [24, 25]. However, there were discrepancies among the results of previous studies, highlighting the importance of assessing gut microbiota composition in populations with different ethnicities.
In this study, we observed higher abundance of Prevotella in diabetic patients and on the contrary higher abundance of Bacteroides in healthy control subjects. Results of previous studies regarding to the bacterial load of Prevotella and Bacteroides in type 1 and type 2 diabetes were controversial [26,27,28]. In contrast to our results, Leiva-Gea et al. nominated Bacteroides as the main genus leading to the gut dysbiosis associated with type 1 diabetes [26]. Besides, Prevotella has conflicting effects on glycemic control. From one side, it is a mucin degrader bacterium disturbing the gut integrity and on the other hand, it is a succinate producer leading to inhibition of hepatic glucose output by stimulation of intestinal gluconeogenesis [26]. However, it should be noted that Prevotella and Bacteroides enterotypes are under influence of long-term dietary intakes. It has been shown that Bacteroides enterotype was associated with a diet rich in protein and animal fat, while the Prevotella enterotype was associated with a diet rich in carbohydrate and fiber [29, 30]. So we can propose that the higher abundance of Prevotella in diabetic patients is a consequence of high carbohydrate intake during patients’ life.
Roseburia as an important butyrate-producing genus could inhibit expression of proinflammatory cytokines in the mucosa and has modulating effects on immune system. Mediterranean diet and fermentable dietary carbohydrates are associated with an increased abundance of intestinal Roseburia and better glycemic control. Evidences have been proposed that the abundance of intestinal Roseburia negatively correlated with plasma glucose, indicating to the role of Roseburia in glucose homeostasis [31]. Consistently, our results revealed lower concentration of Roseburia in faeces of diabetic patients.
In the present study, bacterial load of Faecalibacterium in faeces of type 1 diabetic patients was lower compared to the control group. In agreement with our findings, Huang et al. in a study on young adults observed that abundance of Faecalibacterium correlated negatively with HbA1c levels [32]. Moreover, lower relative abundance of Faecalibacterium has been indicated in type 1 diabetic children [26].
We found that the abundance of Akkermansia was relatively lower in the diabetic groups compared to the healthy subjects; however this difference was not statistically significant. Zhang et al. suggested that low abundance of Akkermansia muciniphila could be a biomarker for glucose intolerance [33]. Furthermore, Endesfelder et al. revealed that alterations in the abundance of mucin-degrading bacteria were associated with early development of islet autoimmunity and type 1 diabetes [34].
Decrease in levels of SCFAs-producing bacteria including Akkermansia, Roseburia and Faecalibacterium which is often observed in gut microbiota of diabetic patients is associated with higher intestinal permeability, allowed translocation of proinflammatory substances from gut lumen to blood circulation causing metabolic endotoxemia [14, 35].
Our results were in line with the studies conducted by Sedighi et al. and Larsen et al. observing meaningful higher concentration of Lactobacillus in type 2 diabetic patients compared to the controls [20, 36]. Ordiz et al. revealed that consumption of dietary starch which is mostly prevalent in diabetic patients changed the gut microbiota composition, with an increase in Lactobacillus genus [37]. However, it should be noted that different species in the same genus could exhibit various properties. For example, Halawa et al. observed lower stool Lactobacillus acidophilus in diabetic patients compared to healthy individuals [38]. Consistent to our findings, colonization of Bifidobacterium with anti-inflammatory effects and bifidogenic properties was lower in gut microbiota of patients with type 1 and type 2 diabetes [26, 36, 39].
Similar to our observation, the abundance of Escherichia belonging to the Proteobacteria phylum has been reported to increase in type 2 diabetic patients [25]. Lipopolysaccharides derived from the outer membranes of Gram-negative bacteria are known to promote secretion of pro-inflammatory cytokines by binding to Toll-like receptor 4 (TLR4) and thus induce insulin resistance. Moreover, increased proportion of proinflammatory bacteria like Escherichia exposed subjects to the impaired epithelial integrity, low-grade inflammation, and autoimmune responses and increased risk of type 1 diabetes [26]. Since evidences showed that medications like metformin and insulin could influence bacterial composition of gut microbiota and may confound the alterations in microbiome, we cannot distinguish between gut microbiota alterations resulting from disease or drug consumption [27, 40, 41]. Metformin treatment has been shown to affect abundances of SCFAs-producing bacteria, Lactobacillus and Escherichia [41]. Moreover, insulin treatment could reverse the observed alterations in Prevotella and Bacteroides abundances in type 1 diabetic patients [27].
Controversial results regarding the gut microbiota composition in type 1 and type 2 diabetes could be partly explained by the heterogeneity in various factors such as ethnicity, geography, medical history, life style and dietary habits of participants, design of studies and methods of gut microbiota assessment. In the present study, we tried to eliminate the effects of some confounders such as antibiotics, prebiotics or probiotics and other chronic disorders by considering them as exclusion criteria. However, since we did not match three groups for sex, age and BMI, we could not remove the potential confounding effects of these factors. Moreover, in the current study we showed alterations of gut microbiota composition in diabetic patients, however, considering the design of this study we could not establish the casual link between microbiome changes and diseases. Further prospective longitudinal studies are warranted to elucidate the cause and effect relationships. Furthermore, we examined only a limited number of bacterial genera in gut microbiota, but a metagenomic assessment can provide a comprehensive view of all microorganisms inhabited in the gastrointestinal tract.
Conclusion
In conclusion, result of this study showed that despite the non-significant difference in Akkermansia level between diabetic patients and healthy subjects, abundance of Escherichia, Prevotella and Lactobacillus was significantly higher in faecal samples of type 1 and type 2 diabetic patients and abundance of Bifidobacterium, Roseburia, Faecalibacterium and Bacteroides was significantly higher in healthy control subjects. By determining the gut microbiota alterations in diabetes, interventional strategies could be designed to modulate the gut microbiota composition and function with the ultimate aim of help in glycemic control.
Data Availability
Data will be provided in case of requesting.
References
Unwin N, Gan D, Whiting D. The IDF diabetes atlas: providing evidence, raising awareness and promoting action. Diabetes Res Clin Pract. 2010;87(1):2–3.
Ejtahed HS, Soroush AR, Angoorani P, Larijani B, Hasani-Ranjbar S. Gut Microbiota as a Target in the Pathogenesis of Metabolic Disorders: A New Approach to Novel Therapeutic Agents. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2016;48(6):349–58.
Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut. 2014;63(9):1513–21.
Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science. 2012;336(6086):1262–7.
Ejtahed HS, Hasani-Ranjbar S, Larijani B. Human microbiome as an approach to personalized medicine. Altern Ther Health Med. 2017;23(6):8–9.
Ejtahed HS, Soroush AR, Siadat SD, Hoseini-Tavassol Z, Larijani B, Hasani-Ranjbar S. Targeting obesity management through gut microbiota modulation by herbal products: a systematic review. Complementary therapies in medicine. 2019;42:184–204.
Ejtahed HS, Angoorani P, Hasani-Ranjbar S, Siadat SD, Ghasemi N, Larijani B, et al. Adaptation of human gut microbiota to bariatric surgeries in morbidly obese patients: a systematic review. Microb Pathog. 2018;116:13–21.
Everard A, Cani PD. Diabetes, obesity and gut microbiota. Best Pract Res Clin Gastroenterol. 2013;27(1):73–83.
Erejuwa O, Sulaiman S, Wahab M. Modulation of gut microbiota in the management of metabolic disorders: the prospects and challenges. Int J Mol Sci. 2014;15(3):4158–88.
Ejtahed HS, Hasani-Ranjbar S. Neuromodulatory effect of microbiome on gut-brain axis; new target for obesity drugs. 2019;18(1):263–5.
Baothman OA, Zamzami MA, Taher I, Abubaker J, Abu-Farha M. The role of gut microbiota in the development of obesity and diabetes. Lipids Health Dis. 2016;15(1):108.
Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes. 2012;3(4):279–88.
Gulden E, Wong FS, Wen L. The gut microbiota and Type 1 Diabetes. Clinical immunology (Orlando, Fla). 2015;159(2):143–53.
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6(10):e25792.
Gérard C, Vidal H. Impact of gut microbiota on host glycemic control. Front Endocrinol. 2019;10:29.
Bartosch S, Fite A, Macfarlane GT, McMurdo ME. Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatment on the fecal microbiota. Appl Environ Microbiol. 2004;70(6):3575–81.
Fitzgerald CB, Shkoporov AN. Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa 2018;19(1):931.
Gregory KE, LaPlante RD, Shan G, Kumar DV, Gregas M. Mode of birth influences preterm infant intestinal colonization with Bacteroides over the early neonatal period. Advances in neonatal care : official journal of the National Association of Neonatal Nurses. 2015;15(6):386–93.
Kanno T, Matsuki T, Oka M, Utsunomiya H, Inada K, Magari H, et al. Gastric acid reduction leads to an alteration in lower intestinal microflora. Biochem Biophys Res Commun. 2009;381(4):666–70.
Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085.
Payne AN, Chassard C, Banz Y, Lacroix C. The composition and metabolic activity of child gut microbiota demonstrate differential adaptation to varied nutrient loads in an in vitro model of colonic fermentation. FEMS Microbiol Ecol. 2012;80(3):608–23.
Rinttila T, Kassinen A, Malinen E, Krogius L, Palva A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004;97(6):1166–77.
Schneeberger M, Everard A, Gómez-Valadés AG, Matamoros S, Ramírez S, Delzenne NM, et al. Akkermansia muciniphila inversely correlates with the onset of inflammation, altered adipose tissue metabolism and metabolic disorders during obesity in mice. Sci Rep. 2015;5:16643.
Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60.
Leiva-Gea I, Sanchez-Alcoholado L, Martin-Tejedor B, Castellano-Castillo D, Moreno-Indias I, Urda-Cardona A, et al. Gut Microbiota Differs in Composition and Functionality Between Children With Type 1 Diabetes and MODY2 and Healthy Control Subjects: A Case-Control Study. 2018;41(11):2385–95.
Mejia-Leon ME, Petrosino JF, Ajami NJ, Dominguez-Bello MG, de la Barca AM. Fecal microbiota imbalance in Mexican children with type 1 diabetes. Sci Rep. 2014;4:3814.
Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 2013;11:46.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–80.
Hjorth MF, Blædel T, Bendtsen LQ, Lorenzen JK, Holm JB, Kiilerich P, et al. Prevotella-to-Bacteroides ratio predicts body weight and fat loss success on 24-week diets varying in macronutrient composition and dietary fiber: results from a post-hoc analysis. Int J Obes. 2019;43(1):149–57.
Tamanai-Shacoori Z, Smida I, Bousarghin L, Loreal O, Meuric V, Fong SB, et al. Roseburia spp.: a marker of health? Future Microbiol. 2017;12:157–70.
Huang Y, Li SC, Hu J, Ruan HB, Guo HM, Zhang HH, et al. Gut microbiota profiling in Han Chinese with type 1 diabetes. Diabetes Res Clin Pract. 2018;141:256–63.
Zhang X, Shen D, Fang Z, Jie Z, Qiu X, Zhang C, et al. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS One. 2013;8(8):e71108.
Endesfelder D, Engel M, Davis-Richardson AG, Ardissone AN, Achenbach P, Hummel S, et al. Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production. Microbiome. 2016;4:17.
Siljander H, Honkanen J, Knip M. Microbiome and type 1 diabetes. EBioMedicine. 2019;46:512–21.
Sedighi M, Razavi S, Navab-Moghadam F, Khamseh ME, Alaei-Shahmiri F, Mehrtash A, et al. Comparison of gut microbiota in adult patients with type 2 diabetes and healthy individuals. Microb Pathog. 2017;111:362–9.
Ordiz MI, May TD, Mihindukulasuriya K, Martin J, Crowley J, Tarr PI, et al. The effect of dietary resistant starch type 2 on the microbiota and markers of gut inflammation in rural Malawi children. Microbiome. 2015;3:37.
Halawa MR, El-Salam MA, Mostafa BM, Sallout SS. The gut microbiome, Lactobacillus acidophilus; relation with type 2 diabetes mellitus. Curr Diabetes Rev. 2019.
Soyucen E, Gulcan A, Aktuglu-Zeybek AC, Onal H, Kiykim E, Aydin A. Differences in the gut microbiota of healthy children and those with type 1 diabetes. Pediatrics international: official journal of the Japan Pediatric Society. 2014;56(3):336–43.
Ejtahed H-S, Tito RY, Siadat S-D, Hasani-Ranjbar S, Hoseini-Tavassol Z, Rymenans L, et al. Metformin induces weight loss associated with gut microbiota alteration in non-diabetic obese women: a randomized double-blind clinical trial. European journal of endocrinology. 2018;1(aop).
Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528(7581):262–6.
Acknowledgements
We are so thankful of all the participants.
Funding
This work was supported by the National Institute for Medical Research Development (NIMAD) (grant number: 940604).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Consent for publication
NA
Ethics approval and consent to participate
Informed consent was obtained from all the participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ejtahed, HS., Hoseini-Tavassol, Z., Khatami, S. et al. Main gut bacterial composition differs between patients with type 1 and type 2 diabetes and non-diabetic adults. J Diabetes Metab Disord 19, 265–271 (2020). https://doi.org/10.1007/s40200-020-00502-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40200-020-00502-7