
Abstract
The phosphate protective film and micro-galvanic corrosion of biological Mg-3Zn-xNd (x = 0, 0.6, 1.2) alloys were investigated by scanning and transmission electron microscopy, quasi-in-situ observation, scanning Kelvin probe force microscopy (SKPFM) and electrochemical tests. The results revealed the Mg–Zn–Nd phases formed in Mg–3Zn alloy contained with Nd. Adding Nd resulted in a significant decline in the cracks of the phosphate protective film and micro-galvanic corrosion of alloys, which were recorded by quasi-in-situ observation. In addition, the Volta potential difference of Mg–Zn–Nd/α-Mg (~ 188 mV) was lower than MgZn/α-Mg (~ 419 mV) and Zn-rich/α-Mg (~ 260 mV), and the corrosion rates of alloys markedly decreased after the addition of 0.6 wt% Nd. The improvement in corrosion resistance of Nd-containing alloys was mainly attributed to the following: (i) the addition of Nd reduced the Volta potential difference (second phases/α-Mg); (ii) the phosphate protective film containing Nd2O3 deposited on the surface of the alloys, effectively preventing the penetration of harmful anions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Mg–Zn alloys are used as bone implant biomaterial because of their excellent biocompatibility, degradability, low elastic modulus, similar density to human bone and good osteoinductivity [1,2,3,4,5]. However, the rapid degradation rate would result in subcutaneous inflammations, and the alloys’ mechanical integrity was also compromised before the new bone tissue healed [6,7,8]. Therefore, improving the corrosion-resistant performance of Mg–Zn alloys is essential to expand their application in the medical field.
The alloying method has recently been utilized to slow down biological Mg–Zn alloys’ degradation rate. Jiang et al. [9] claimed that after adding 1.5 wt% Sn in the Mg-4Zn alloys, the icorr decreased from 66.4 to 10.6 μA/cm2 in the phosphate buffer solution environment. Cai et al. [10] revealed the formation of the Mg60Zn32Nd8 phase with a lower Volta potential in the Mg–Zn–Zr–(Nd) alloy, thereby reducing the extent of corrosion. The work of Zhang et al. [11] showed that a continuous dense film was formed in Kokubo’s solution environment, which significantly reduced corrosion susceptibility of the Mg–Zn–Mn–(0.6Nd) alloy, improved self-healing ability, and reduced corrosion factors’ adsorption, diffusion, and penetration into the membrane structure. In addition, Cho et al. [12] reported that adding Mn formed extra MnO and MnO2 films in the corrosion film with a depth of 182 nm in Hank’s solution, which effectively inhibited chloride ion penetration. However, the influence of alloying on the film formation of biological Mg–Zn alloys is still unclear, especially in the body fluid environment.
Quasi-in-situ observations were utilized to monitor the corrosion process of alloys in an electrolyte environment, which helped reveal the information on corrosion film [13,14,15,16]. Jin et al. [17] studied the corrosion initiation and development of Mg–0.5Zn–0.2Ca alloy by quasi-in-situ electron microscopic observations. The results showed that the Mg2Ca phase was preferentially corroded as an anodic phase at the early immersion stage, whereas the Ca2Mg6Zn3 and MgCaSi phases were effective cathodes that remained unchanged even after 24 h of immersion. Yang et al. [18] observed that the Mn particle was oxidized in the OCP environment, forming relatively dense Mn-rich films based on the quasi-in-situ scanning electron microscopy (SEM) observations. In addition, quasi-in-situ optical images of Mg–6Mn alloy potentially polarized at − 2 VSCE for 10 and 1200 min showed that the strength of hydrogen flow strength of the alloy decreased significantly after 1200 min immersion in NaCl solutions, indicating the protective effect of the Mn-rich corrosion films.
Moreover, scanning Kelvin probe force microscopy (SKPFM) could measure the Volta potential difference between the second phase and substrate [19,20,21,22], which helps understand the corrosion mechanism. The work of Yin et al. [23] showed that the potential difference between the Al–Mn–Nd/α-Mg and Al2Nd/α-Mg phases was lower than the Al–Mn/α-Mg in the Nd-containing AZ91 alloys, which implied that generation of new second phase attenuated the micro-galvanic corrosion. Jiang et al. [24] revealed that the MgxZny cathode phase had 86 ± 26 mV higher potentials than the α-Mg in the Mg–4Zn–0.2Sn alloys, leading to severe localized corrosion behaviour. Therefore, the quasi-in-situ observations and SKPFM techniques are critical in exploring the corrosion mechanism of biological Mg–Zn alloys.
Previous studies have shown that small amounts of Nd represent good biological safety and the ability to improve the anticorrosion property of Mg–Zn alloys [25,26,27,28]. Niu et al. [29] found that screws made of Mg–Nd–Zn–Zr were degraded in the biological mandibles for 18 months, Mg12Nd particles were degraded, and the interfacial analysis of the implants revealed good osteointegration. In addition, the corrosion current density of the Mg–2Zn–0.2Mn–(Nd) alloys was remarkably reduced by an order of magnitude by adding 0.6 wt% Nd [30]. However, the rationale behind the enhanced corrosion resistance of Mg–Zn alloys by adding Nd remains unclear.
Therefore, this study applied quasi-in-situ observations and SKPFM techniques to study the corrosion behaviour of biological Mg–3Zn–xNd (x = 0, 0.6, 1.2) alloys. Quasi-in-situ optical microscopy combined with SEM observations revealed the corrosion process of the alloys with and without adding Nd in simulated body fluids (SBF). In addition, the SKPFM was used to study the Volta potential difference of the second phase/matrix. Combining the two techniques with traditional corrosion testing can further understood the effects of phosphate protective film and surface micro-galvanic corrosion on biological Mg–3Zn–xNd (x = 0, 0.6, 1.2) alloys in the simulated body fluid environment.
2 Experimental
2.1 Materials preparation
As-cast Mg–3Zn–xNd alloys (x = 0, 0.6, 1.2) were prepared using pure Mg (99.8 wt%), pure Zn (99.9 wt%) and Mg-25 wt% Nd master alloys. The raw materials were melted at 750 °C under a protective gas (Ar) atmosphere. Then, the molten alloys were held for 10 min at 750 °C to homogenize. Finally, the melts were poured into a mould (Ф20 mm × 150 mm), preheated at 200 °C and cooled naturally in the air. The practical chemical analysis of the Mg–3Zn–xNd alloys was obtained by inductively coupled plasma atomic emission spectroscopy (ICP-AES), and the corresponding results are listed in Table 1.
2.2 Microstructural characterization
After grinding by successively SiC paper (up to 2000 grits) and polishing with diamond polishing paste prior, the specimens were corroded with 4 vol.% nital for 5 s, then cleaned with alcohol and air dried. The morphologies of the Mg–3Zn–xNd were observed by scanning electron microscopy (SEM, Quanta 200FEG).
Transmission electron microscopy (TEM, Talos F200X) was used to reveal the composition of the second phase. The samples were ground to 80–100 μm thickness. Next, the discs with a diameter of 3 mm were punched out from the samples and cleaned with alcohol and deionized water for 5 min. Then, the disc was placed on the pit grinder (PIPS 656, GATAN) to get a central pit whose thickness was 10–30 μm. Finally, the thin area of ten nanometers was obtained using a precision ion thinning instrument (PIPS 691, GATAN).
The topographic and Volta potentials of the second phase were gained by SKPFM (Agilent 5500) performing in the tapping mode. The mechanical drive frequency and tip bias were set to 75 kHz and ± 0.05 V, respectively. The distance between the silicon probe with a thickness of 20 nm and the sample was set to 100 nm. All measurements were carried out at room temperature.
2.3 Quasi-in-situ corrosion observation
Quasi-in-situ observations were carried out using SEM and optical microscopy (OM, Nikon MA200) on the same location of specimens immersed in SBF at different times. All samples were prepared according to the sample mentioned above preparation requirements for microscopic observation. The cross marks were made on the sample’s surface to facilitate the identification of the same position before and after corrosion. Firstly, the observation area was selected by SEM, and the second phase with specific characteristics was marked and located. Subsequently, the alloys were immersed in SBF for a specified period, rinsed with ethanol, and dried in cold air. The marked position’s corrosion morphologies were observed and recorded by OM. Finally, SEM was utilized to study the change in morphologies and components of the marked position before and after corrosion.
2.4 Electrochemical test
All electrochemical tests were conducted in SBF solution using the electrochemical workstation (Princeton P4000) equipped cell at 37 °C. The chemical composition of SBF is listed in Table 2. The three-electrode setup consists of a platinum plate (counter electrode), a saturated calomel (reference electrode) and a sample with a contact area of 1 cm2 (working electrode). The samples are placed in SBF solution for 10 min to achieve a steady-state open-circuit potential (OCP). The potentiodynamic polarization test was carried out at a scanning rate of 1 mV s−1 with a scan range of ± 250 mV versus OCP. Furthermore, the electrochemical impedance spectroscopy (EIS) data were obtained from 100 kHz to 0.01 Hz with a perturbed signal of 5 mV. All tests were repeated 5 times to minimize errors.
The values of inhibition efficiency were obtained by the corrosion current density according to the following equation [31]:
where icorr, 0 represents the corrosion current density of the matrix alloy, icorr is defined as the corrosion current density of the modified alloy.
2.5 Hydrogen evolution and weight loss tests
The hydrogen evolution and weight loss were evaluated in SBF solution at 37 °C for 24 h. H2 was collected into the inverted burette above the inverted funnel connection. The samples were immersed in a mixture of 66 g/L CrO3 and 3.3 g/L AgNO3 for 15 min to clear the corrosion products. After that, samples were again gently washed with ethanol and distilled water. Weight loss was measured before and after immersion, and the mean of the experimental data was recorded. The hydrogen evolution and weight loss rates were calculated using the following formula [32]:
where W1 (g) and W2 (g) are the initial and final masses, H1 (ml) and H2 (ml) are the initial liquid level and post-soaking liquid level, A (cm2) and T (day) represent the exposed surface area of specimens and immersion time, respectively.
2.6 Corrosion product analysis
The element distribution on the sample immersed in the SBF solution for 24 h was characterized using X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi). The X-ray source was an X-ray tube with Al as the anode, and its photon energy was 1486 eV. In addition, the scanning range was 0–1300 eV, with a pass-through energy setting of 23.5 eV. The C1s peak (284.8 eV) was used to standardize the binding energy during the analysis. The chemical composition of corrosion products was also analysed by X-ray diffraction (XRD, D8 ADVANCE).
3 Results
3.1 Microstructural
Figure 1 shows the SEM images of the Mg–3Zn–xNd (x = 0, 0.6, 1.2) alloys. As shown in Fig. 1a, the Mg–3Zn alloy consisted of the α-Mg, coarse continuous network of Zn-rich and granular and strip MgZn phases. With increasing Nd content in the alloys, the bright white second phases gradually increased, as shown in Fig. 1b and c. Meanwhile, the morphologies of the bright white second phases changed from dots and strips to a semi-continuous network. Based on the corresponding energy-dispersive spectroscopy (EDS) mapping of Mg–3Zn–0.6Nd alloy and Mg–3Zn–1.2Nd alloy in Fig. 2, the bright white second phase was enriched in both Zn and Nd.

SEM results of Mg–3Zn–xNd alloys: a Mg–3Zn alloy; b Mg–3Zn–0.6Nd alloy; c Mg–3Zn–1.2Nd alloy

Phase morphologies and the corresponding EDS mapping results of Mg–3Zn–0.6Nd alloy a–d and Mg–3Zn–1.2Nd alloy e–i
TEM analysis further detected the second phase compositions of the Mg–3Zn–1.2Nd alloy, as shown in Fig. 3a–d. Nd and Zn elements are enriched in the round-like black particles, as shown in Fig. 3a and b. Furthermore, Fig. 3c and d shows that the needle-shaped particle with a 1.5 μm diameter was identified as an Mg78Zn0.56Nd20 ternary phase based on the atomic ratios.

TEM images a, c of Mg–3Zn–1.2Nd alloy and corresponding EDS analysis b, d
Figure 4a–d presents the surface topography, surface potential distribution and potential profile of the Mg–3Zn alloys. The α-Mg matrix, white MgZn particles and coarse net-like Zn-rich phases were observed in the matrix alloy (Fig. 4a and b), which conforms to the SEM results in Fig. 1. The Volta potentials difference of the MgZn phase and α-Mg was ~ 419 mV, whereas that of the Zn-rich phase was ~ 260 mV relative to the α-Mg phase, as exhibited in Fig. 4c and d. Therefore, the MgZn and Zn-rich phases revealed more positive Volta potentials relative to the α-Mg matrix, suggesting cathodic activity. Hence, the corrosion occurred preferentially at the interface between the MgZn/Zn-rich phases (cathodes) and α-Mg matrix (anode).

2D surface topography, 3D surface topography, 3D surface potential map and the relative potential curve on the A–B line obtained from SKPFM results of Mg–3Zn alloy a–d and Mg–3Zn–0.6Nd alloy e–h
Figure 4e–h displays the surface topography, surface potential distribution and potential profile of the Mg–3Zn–0.6Nd alloy. The line-profile analyses from A to B in Fig. 4i show that the Mg–Zn–Nd phase was ~ 188 mV than that of the α-Mg matrix, indicating the Volta potential difference of the second phases was significantly reduced with the addition of Nd. Thus, it can be confirmed that the Mg–Zn–Nd phase was less cathodic than the MgZn and Zn-rich phases, indicating that the driving force of micro-galvanic corrosion was significantly weakened.
3.2 Quasi-in-situ corrosion observation
Figure 5 exhibits the optical microscope images of the quasi-in-situ corrosion morphology of Mg–3Zn–xNd alloys immersed in SBF at different times. As shown in Fig. 5a, the MgZn phase was marked, and then the quasi-in-situ corrosion process of these MgZn phases was observed. After being immersed for the 30 s, corrosion occurred at the interface of the MgZn phase/α-Mg matrix, as shown by the circled area in Fig. 5b. Then, the corrosion to the α-Mg matrix diffused outward along the interface at the 60 s (Fig. 5c). When the soaking time increased to 180 s, the surroundings of the MgZn phase completely turned black, indicating severe corrosion in the matrix.

Optical microscope images of quasi-in-situ corrosion morphology of Mg–3Zn–xNd (x = 0, 0.6, 1.2) alloys: a–d Mg–3Zn alloy; e–h Mg–3Zn–0.6Nd alloy; i–l Mg–3Zn–1.2Nd alloy
Quasi-in-situ corrosion morphology changes in the Mg–3Zn–0.6Nd alloy are shown in Fig. 5e–h. At the onset of corrosion, slight corrosion mainly occurred on the substrate around the Mg–Zn–Nd phase. At the same time, the majority of the α-Mg matrix remained unchanged, as shown in Fig. 5f. The interface corrosion at the Mg–Zn–Nd phase and substrate was gradually deepened when the immersion time increased from the 30 to 60 s. In contrast, the corrosion had not spread significantly from the 60 to 180 s, as shown in Fig. 5g and h. In addition, Fig. 5i–l shows the quasi-in-situ corrosion morphology of Mg–3Zn–1.2Nd alloy. In the initial stage of corrosion, the matrix near the second phase was gradually oxidized. When the corrosion process continued to the 180 s, part of the matrix oxidation degree deepened and turned black.
Figure 6a–d shows the SEM images and the corresponding EDS mapping of the Mg–3Zn alloy before and after corrosion. At the same delineated position as in the optical microscope images, the MgZn phase with specific morphological characteristics can be observed (Fig. 6a and b). It is worth noting that many cracks existed in the corrosion film on the surface of the alloy after corrosion (Fig. 6c). Furthermore, EDS mapping results show that the O and P elements are evenly distributed on the surface of the alloy, suggesting that the main component of the corrosion film is phosphate (Fig. 6d).

SEM images of the quasi-in-situ corrosion observation area and the corresponding EDS mapping of the Mg–3Zn–xNd (x = 0, 0.6, 1.2) alloys before and after corrosion: a–d Mg–3Zn alloy; e–h Mg–3Zn–0.6Nd alloy; i–l Mg–3Zn–1.2Nd alloy
The SEM images and the corresponding EDS mapping of the Mg–3Zn–0.6Nd alloy before and after corrosion are shown in Fig. 6e–h. In Fig. 6e and f, the Mg–Zn–Nd phase can be observed in the circled positions corresponding to the optical microscope images. After the quasi-in-situ corrosion, the cracks of the surface film of the Mg–3Zn–0.6Nd alloy are less than the matrix alloy, which means that a comparatively stable film exists on the corrosion surface. Figure 6i–l also shows the marked Mg–Zn–Nd phase of the Mg–3Zn–1.2Nd alloy before and after corrosion. The second phase is distributed in a network structure, and there are also cracks on the surface of the alloy after corrosion, as shown in the delineation position.
3.3 Electrochemical test
The polarization curves of the alloys soaked in SBF at 37 °C for 0 and 24 h are displayed in Fig. 7. The fitting results of Tafel curves are listed in Table 3. The Mg–3Zn alloy showed the lowest corrosion potential (Ecorr = − 1.789 VSCE) and maximum corrosion current density (icorr = 276.8 μA/cm2), as shown in Fig. 7a. Moreover, a significant positive shift of the corrosion potential for Mg–3Zn alloys modified by Nd, the Ecorr value of Mg–3Zn–0.6Nd and Mg–3Zn–1.2Nd was − 1.619 and 1.635 VSCE, respectively. Meanwhile, the icorr decreased from 276.8 to 140.042 μA/cm2 after adding 0.6 wt% Nd, while the icorr of Mg–3Zn–1.2Nd decreased to 180.548 μA/cm2. After 24 h of soaking in the SBF solution (Fig. 7b), the icorr value decreased by an order of magnitude after adding 0.6 wt% Nd (from 157.805 to 12.35 μA/cm2). Noticeably, the Ecorr value shifted to a more noble value (− 1.131 VSCE), implying a weakening corrosion tendency [33]. In addition, Table 4 further lists the inhibition efficiency of the alloy and summarizes the inhibition efficiency results obtained by other researchers for the Nd-modified alloys, which indicates that the Mg–3Zn–0.6Nd alloy shows a good inhibition efficiency after 24 h of immersion [10, 11, 27, 28, 30].

Potentiodynamic polarization curves of Mg–3Zn–xNd(x = 0, 0.6, 1.2) soaked in SBF at 37 °C: a 0 h; b 24 h
Figure 8 shows the EIS test results of the alloys in SBF and the equivalent fitting circuit of the EIS. All the Nyquist plots have a high frequency and a medium frequency capacitive arc (Fig. 8a). The diameter of a capacitive arc in the high frequency range is equivalent to the surface charge transfer resistance of the alloy [34,35,36]. Therefore, the corrosion rate can be evaluated by the diameter of the capacitance reactance arc, which was ranked as Mg–3Zn–0.6Nd > Mg–3Zn–1.2Nd > Mg–3Zn, is consistent with the results of the potentiodynamic polarization curve. After immersion treatment in SBF at 37 °C for 24 h, the diameter of the capacitance reactance arc of the samples all increased, as shown in Fig. 8b. Among them, the improvement of electrochemical impedance parameters of the Mg–3Zn–0.6Nd alloy is much larger than that of other alloys.

EIS test results of Mg–3Zn–xNd (x = 0, 0.6, 1.2) immersion in SBF and equivalent fitting circuit of the EIS test: a 0 h; b 24 h; c equivalent fitting circuit
Figure 8e and Table 5 show the equivalent circuit model and fitting parameter results of the EIS test results of the Mg–3Zn–xNd (x = 0, 0.6, 1.2) alloys. Rs is the solution resistance. Rct is related to resistance to transfer charge. Rf and CPEf represent corrosion product layer resistance and capacitance, respectively. CPEdl represents the capacitance of the double layer between the alloy and the corrosive medium [37,38,39]. The fitted electrochemical parameters show that the polarization resistance of the modified alloy increases. The Rct of the Mg–3Zn–0.6Nd alloy before and after immersion is 145.5 and 4271 Ω cm2, respectively, which is higher than that of other alloys.
3.4 Hydrogen evolution and weight loss tests
Figure 9 shows the hydrogen evolution volume and corrosion rates calculated from weight loss of Mg–3Zn–xNd (x = 0, 0.6, 1.2) alloys after being immersed in SBF for 24 h. As shown in Fig. 9a, the addition of 0.6 wt% Nd effectively reduces the rate of hydrogen evolution, and the value was about 2.885 mL cm−2 d−1. Further increases in Nd content increased the volume of hydrogen evolution slightly, but still less than that of the matrix alloy. According to the result of weight loss tests (Fig. 9b), the corrosion rate of Mg–3Zn alloy was significantly inhibited by adding element Nd, and Mg–3Zn–0.6Nd represented the lowest weight loss value of 3.23 ± 0.31 mg cm−2 d−1. However, the corrosion rate of the alloy increased slightly when the Nd content increased to 1.2 wt%.

Hydrogen evolution volume a and weight loss rates b of Mg–3Zn–xNd (x = 0, 0.6, 1.2) alloys after immersed in SBF for 24 h
3.5 Corrosion morphology and product analysis
Figure 10 shows the corrosion morphology of the Mg–3Zn and Mg–3Zn–0.6Nd alloys immersed in SBF for 24 h. It can be observed that there are large corrosion pits on the surface of the matrix alloy (Fig. 10a). Combined with the EDS results, it can be seen that phosphorus is enriched in the uncorroded area, as shown in Fig. 10b. In contrast, only a few pitting sites are formed on the Mg–3Zn–0.6Nd alloy, and P is evenly distributed on the surface (Fig. 10c and d). In addition, there are more pitting pits on the Mg–3Zn–1.2Nd alloy, and EDS results show that a large area of magnesium substrate is exposed, as shown in Fig. 10e and f.

Surface corrosion morphology images and corresponding EDS mapping of tested alloys immersed in SBF for 24 h: a, b Mg–3Zn alloy; c, d Mg–3Zn–0.6Nd alloy; e, f Mg–3Zn–1.2Nd alloy
The longitudinal images and the corresponding EDS mapping of the tested alloys were characterized and shown in Fig. 11. Figure 11a reveals that a corrosion pit with a depth of ~ 92 μm was formed inside the matrix alloy, whereas a corrosion film of thickness ~ 15 μm can be observed on the corresponding longitudinal view of the Mg–3Zn–0.6Nd alloy, as shown in Fig. 11b. Figure 11c shows that a shallow corrosion pit was formed within the Mg–3Zn–1.2Nd alloy, with a depth of ~ 38 μm. There was a relatively dense corrosion film on the surface of the Mg–3Zn–0.6Nd alloy, which hindered the further diffusion of corrosion. According to the local magnification image and the corresponding EDS mapping result, a phosphating film was enriched on the surface of the alloy (Fig. 11d and e).

Longitudinal images and the corresponding EDS mapping of tested alloys immersed in SBF for 24 h: a Mg–3Zn alloy; b Mg–3Zn–0.6Nd alloy; c Mg–3Zn–1.2Nd alloy; d locally enlarged longitudinal image; e EDS result
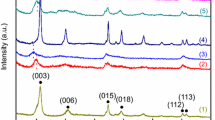
To further explore the corrosion products, XRD and XPS analyses were performed for Mg–3Zn and Mg–3Zn–0.6Nd alloy after immersion in SBF for 24 h, as shown in Figs. 12 and 13. The corrosion products of the alloys are mainly composed of MgCO3·3H2O, Mg(OH)2, (CaMg)x(PO4)y. The XPS pattern and fitting results of the corrosion products of the Mg–3Zn–0.6Nd alloy are shown in Fig. 13. The peak centres of Ca 2p1 and Ca 2p3 are located at 350.7 and 347.1 eV (Fig. 13a), respectively, representing the presence of \({\text{Ca}} - {\text{CO}}_{{3}}^{{{2} - }}\) and \({\text{Ca}} - {\text{PO}}_{{4}}^{{{3} - }}\) in the corrosion products [40, 41]. O 1s spectrum mainly contains three peaks of C–O, O–H and \({\text{CO}}_{{3}}^{{{2} - }}\) (Fig. 13b). The fitting results in the P 2p spectrum show that two of the peaks are and \({\text{H}}_{{2}} {\text{PO}}_{{4}}^{{{3} - }}\), respectively (Fig. 13c) [42, 43]. Moreover, the peak centres of Nd 3d3 and Nd 3d5 are located at 1002.9 and 982.4 eV, indicating that Nd2O3 is contained in the corrosion products [44].

XRD results of corrosion products produced by test samples (Mg–3Zn, Mg–3Zn–0.6Nd) after immersion in SBF for 24 h

XPS analysis of the surface corrosion films on Mg–3Zn–0.6Nd alloy: a Ca 2p region; b O 1s region; c P 2p region; d Nd 3d region
4 Discussion
4.1 Mechanism of phosphate protective film
Generally, the biological magnesium alloys tend to form a phosphorus-containing corrosion film in body fluids, which is considered an effective barrier against corrosion factors such as Cl−, HPO42− and HCO3− in body fluids [45]. According to XRD and XPS results of corrosion products, the Mg–3Zn–xNd alloys were covered with a phosphate film, which was due to the following reactions that occurred in the SBF solution:
The Tafel and EIS results show that the corrosion performance of the alloys after immersion for 24 h was significantly improved, which implied that the (CaMg)x(PO4)y protective film could act as an effective barrier to prevent ion exchange between solute and solution to a certain extent [46].
The micro-galvanic corrosion caused many cracks in the matrix alloy’s phosphate protective film, according to the quasi-in-situ observation results in Fig. 6c. The micro-galvanic corrosion would produce a large amount of hydrogen quickly and destroy the formation of a phosphate protective film. In contrast, only a few surface cracks on the surface of the alloy contained with Nd after immersion were observed in Fig. 6g, which benefited from the formation of the relatively weaker micro-galvanic corrosion. The slow dissolution rate will significantly weaken the destructive effect of hydrogen on the phosphate protective film and allow the corrosion products to accumulate more effectively on the surface of the alloy.
In addition, Nd2O3 also plays a vital role in the phosphate protective film. Pilling-Bedworth ratio (PBR) is usually used to evaluate the stability of oxide film, and the relevant equation is as follows [47]:
where \(V_{{{\text{comp}}}} /V_{{\text{M}}}\) is the volume ratio between oxide and metal, and \(\rho_{\text{M}}\) and \(\rho_{{{\text{comp}}}}\) denote corresponding densities, respectively. \(M_{{{\text{comp}}}}\) and n are the molar mass and the atom numbers of the oxide, and A denotes the relative atomic mass of the metal. Usually, the film is dense and protective when the PBR is between 1 and 2. Moreover, the PBR value of Nd2O3 is 1.13 (> 1), indicating that the phosphate protective film containing Nd2O3 is compact [48]. It serves as a physical barrier to precipitate on the defects, thereby further increasing the inhibition of ion exchange by the phosphate protective film.
4.2 Effect of micro-galvanic corrosion and corrosion mechanism of Mg–3Zn–xNd alloys
In order to convey the joint influence of phosphate protective film and micro-galvanic corrosion, the schematic diagrams are shown in Fig. 14. In the matrix alloy, two kinds of micro-galvanic corrosion (MgZn/α-Mg and Zn-rich/α-Mg) were formed, as shown in Fig. 14a. Moreover, the micro-galvanic corrosion occurs near the MgZn phase due to the MgZn phase (~ 419 mV) as the primary cathode and the zinc-rich phase (~ 260 mV) as the secondary cathode. During corrosion, the anodic magnesium matrix with low potential next to the second phase undergoes a redox reaction with water molecules to dissolve (\({\text{Mg}} \to {\text{Mg}}^{2 + } + 2{\text{e}}^{ - }\) and \(2{\text{H}}_{2} {\text{O}} + 2{\text{e}}^{ - } \to 2{\text{OH}}^{ - } + {\text{H}}_{2} \uparrow\)). Due to the Ksp (solubility product constant) of phosphate is much lower than that of Mg(OH)2 (The Ksp(Mg(OH)2) = 5.61 × 10−12, while the value of calcium magnesium phosphate on the order of 10−28), OH− is released and preferentially participates in the formation of phosphate film [49, 50]. However, severe micro-galvanic corrosion leads to the failure of phosphate to form a barrier in the corrosion pit in time, weakening the phosphate film’s protective effect (Fig. 14b).

Corrosion mechanism of Mg–3Zn–xNd (x = 0, 0.6,1.2) alloys in SBF: a, b Mg–3Zn alloy; c, d Mg–3Zn–xNd alloy
When Nd was added into the Mg–3Zn alloy, two micro-galvanic couples (Mg–Zn–Nd/α-Mg and Zn-rich/α-Mg) existed in the Mg–3Zn–xNd alloys, as shown in Fig. 14c. The formation of the low-potential Mg–Zn–Nd phase (~ 188 mV) consumes part of the Zn element and inhibits the formation of the high-potential MgZn phase and Zn-rich phase, which makes the micro-galvanic corrosion weaker. In addition, the combined results of electrochemical tests and weight loss experiments revealed that the Mg–3Zn–0.6Nd alloy showed a lower icorr and weight loss rate than the matrix alloy, which confirms the mitigation of micro-galvanic corrosion. Furthermore, owing to the low degradation rate of the alloy in the SBF, the corrosion product can be effectively deposited on the alloy surface. Moreover, Nd2O3 will fill the cracks and defects of the film, forming a uniform layer of phosphate protective film (Fig. 14d). Finally, the resistance of the Mg–3Zn–0.6Nd alloy to corrosion factors is significantly improved. However, as the content of Nd increases to 1.2 wt%, the second phase in the Mg–3Zn–1.2Nd alloy increases and the morphology changes to a semi-continuous network. Due to the potential difference between the second phases and the matrix, the increase in the second phases will more easily lead to micro-galvanic corrosion, which reduces the corrosion resistance of the Mg–3Zn–1.2Nd alloy. Meanwhile, the film-forming properties of the Mg–3Zn–1.2Nd alloy surface will be affected, resulting in more exposed magnesium substrate area on the surface, as shown in Fig. 10e and f.
5 Conclusions
-
1.
The addition of Nd in Mg–3Zn alloy led to the formation of Mg–Zn–Nd phases, resulting in the morphology of the second phase shift from granular and strip to a semi-continuous network structure.
-
2.
The results of SKPFM showed that the Volta potential values of Mg–Zn–Nd/α-Mg (~ 188 mV) were significantly lower than that of the MgZn/α-Mg (~ 419 mV) and Zn-rich/α-Mg (~ 260 mV).
-
3.
Quasi-in-situ observation indicated that the addition of Nd significantly densified the phosphate protective film and weakened the effect of micro-galvanic corrosion.
-
4.
The phosphate protective film containing Nd2O3 deposited on the surface of the alloys, effectively preventing the penetration of harmful anions.
-
5.
The Mg–3Zn–0.6Nd alloy exhibited the lowest icorr of 140.042 μA/cm2 and the highest Rct of 145.5 Ω cm2, suggesting superior corrosion resistance.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also form part of an ongoing study.
References
V.C. Shunmugasamy, M. AbdelGawad, M.U. Sohail, T. Ibrahim, T. Khan, T.D. Seers, B. Mansoor, Bioact. Mater. 28, 448 (2023)
C.J. Yan, B. Guan, Y.C. Xin, L.Y. Zhao, G.J. Huang, R. Hong, X.B. Chen, P.K. Chu, Acta Metall. Sin. -Engl. Lett. 36, 439 (2023)
Z. Cui, W. Li, L. Cheng, D. Gong, W. Cheng, W. Wang, Mater. Charact. 151, 620 (2019)
M. Hashemi, R. Alizadeh, T.G. Langdon, J. Magnes. Alloy. 11, 7 (2023)
L. Shi, Y. Yan, C.S. Shao, K. Yu, B. Zhang, L.J. Chen, J. Magnes. Alloy. (2022)
S. Cai, T. Lei, N. Li, F. Feng, Mater. Sci. Eng. C 32, 2570 (2012)
S. Jafari, R.S. Raman, C.H. Davies, J. Hofstetter, P.J. Uggowitzer, J.F. Löffler, J. Mech. Behav. Biomed. Mater. 65, 634 (2017)
S.E. Harandi, P.C. Banerjee, C.D. Easton, R.S. Raman, Mater. Sci. Eng. C 80, 335 (2017)
W. Jiang, J. Wang, W. Zhao, Q. Liu, D. Jiang, S. Guo, J. Magnes. Alloy. 7, 15 (2019)
C. Cai, R. Song, L. Wang, J. Li, Appl. Surf. Sci. 462, 243 (2018)
Y. Zhang, J. Li, J. Li, J. Alloys Compd. 730, 458 (2018)
D.H. Cho, B.W. Lee, J.Y. Park, K.M. Cho, I.M. Park, J. Alloys Compd. 695, 1166 (2017)
Y. Cubides, A.I. Karayan, D. Zhao, L. Nash, K. Xie, H. Castaneda, J. Alloys Compd. 840, 155786 (2020)
J. Shen, T. Lai, Z. Yin, Y. Chen, K. Wang, H. Yan, H. Song, R. Liu, C. Luo, Z. Hu, J. Magnes. Alloy. (2022). https://doi.org/10.1016/j.jma.2022.06.016
R. Ambat, N.N. Aung, W. Zhou, Corros. Sci. 42, 1433 (2000)
J. Kwon, S.-M. Baek, H. Jung, J.C. Kim, S.Y. Lee, S.S. Park, Corros. Sci. 185, 109425 (2021)
Y. Jin, C. Blawert, F. Feyerabend, J. Bohlen, M.S. Campos, S. Gavras, B. Wiese, D. Mei, M. Deng, H. Yang, Corros. Sci. 158, 108096 (2019)
L. Yang, S. He, C. Yang, X. Zhou, X. Lu, Y. Huang, G. Qin, E. Zhang, J. Magnes. Alloy. 9, 676 (2021)
T. Lai, Z. Yin, J. Shen, P. Sun, Q. Liu, K. Wang, H. Yan, H. Song, C. Luo, Z. Hu, J. Electrochem. Soc. 168, 081502 (2021)
R. Jia, M. Zhang, L. Zhang, W. Zhang, F. Guo, J. Alloys Compd. 634, 263 (2015)
R. Arrabal, A. Pardo, M. Merino, M. Mohedano, P. Casajús, K. Paucar, G. Garcés, Corros. Sci. 55, 301 (2012)
K.H. Anantha, C. Örnek, S. Ejnermark, A. Medvedeva, J. Sjöström, J. Pan, J. Electrochem. Soc. 164, C85 (2017)
Z. Yin, R. He, Y. Chen, Z. Yin, K. Yan, K. Wang, H. Yan, H. Song, C. Yin, H. Guan, Appl. Surf. Sci. 536, 147761 (2021)
P. Jiang, C. Blawert, N. Scharnagl, J. Bohlen, M.L. Zheludkevich, Corros. Sci. 174, 108863 (2020)
B. Venkateswarlu, B.R. Sunil, R.S. Kumar, Mater. Today Proc. (2023). https://doi.org/10.1016/j.matpr.2023.02.342
D.Y. Ding, Y.H. Du, M.F. Tang, S. Bo, G. Ning, H.J. Zhang, S.F. Guo, Trans. Nonferr. Metal. Soc. 33, 7 (2023)
Z. Cao, G. Kong, C. Che, Y. Wang, Appl. Surf. Sci. 426, 67 (2017)
B. Mingo, R. Arrabal, M. Mohedano, C. Mendis, R. Del Olmo, E. Matykina, N. Hort, M. Merino, A. Pardo, Mater. Des. 130, 48 (2017)
J. Niu, M. Xiong, X. Guan, J. Zhang, H. Huang, J. Pei, G. Yuan, Corros. Sci. 113, 183 (2016)
Y. Zhang, J.Y. Li, P.K. Liaw, Y.Z. Xu, H.Y. Lai, J. Alloys Compd. 769, 552 (2018)
Y. Chen, X. Wang, T. Lai, D. Liu, J. Pan, L. Lin, H. Guan, C. Luo, H. Song, Y. Xin, J. Mol. Liq. 357, 119095 (2022)
Z. Hu, Z. Yin, Z. Yin, K. Wang, Q. Liu, P. Sun, H. Yan, H. Song, C. Luo, H. Guan, Corros. Sci. 176, 108923 (2020)
Z. Hu, R. Liu, S. Kairy, X. Li, H. Yan, N. Birbilis, Corros. Sci. 149, 144 (2019)
X.Z. Shi, Z.Y. Cui, J. Li, B.C. Hu, Y.Q. An, X. Wang, H.Z. Cui, Acta Metall. Sin. -Engl. Lett. 36, 1421 (2023)
X. Yan, P. Wan, L. Tan, M. Zhao, L. Qin, K. Yang, Mater. Sci. Eng. C 93, 565 (2018)
T. Bu, R. Jia, T. Ying, A. Atrens, P. Chen, D. Zheng, F. Cao, Acta Metall. Sin. -Engl. Lett. 36, 1630 (2023)
S. Mohajernia, S. Hejazi, A. Eslami, M. Saremi, Surf. Coat. Technol. 263, 54 (2015)
Y. Song, E.-H. Han, D. Shan, C.D. Yim, B.S. You, Corros. Sci. 65, 322 (2012)
N. Dinodi, A.N. Shetty, J. Magnes. Alloy. 1, 201 (2013)
J. Chen, T. Jiang, X. Lan, K. Kang, F. Cai, S. Ma, Mater. Today Phys. 20, 100474 (2021)
Z. Xu, H. Liu, G. Hu, X. Zhuo, K. Yan, J. Ju, W. Wang, H. Teng, J. Jiang, J. Bai, Acta Metall. Sin. -Engl. Lett. 36, 1833 (2023)
X. Cui, Q. Li, Y. Li, F. Wang, G. Jin, M. Ding, Appl. Surf. Sci. 255, 2098 (2008)
Y. Song, D. Shan, R. Chen, F. Zhang, E.-H. Han, Surf. Coat. Technol. 203, 1107 (2009)
C.H. Kao, T. Chan, K.S. Chen, Y.T. Chung, W.S. Luo, Microelectron. Reliab. 50, 709 (2010)
Z.F. Al-Sherify, N.M. Dawood, Z.T. Khulief, Mater. Today: Proc. 61, 1100 (2022)
T. Lei, W. Tang, S.H. Cai, F.F. Feng, N.F. Li, Corros. Sci. 54, 270 (2012)
R.C. Zeng, L. Sun, Y.F. Zheng, H.Z. Cui, E.H. Han, Corros. Sci. 79, 69 (2014)
Y. Song, Y.H. Liu, S. Yu, X. Zhu, S. Wang, J. Mater. Sci. 42, 4435 (2007)
S. Zhong, C. Wu, J. Li, Y. Chen, Y. Wang, Y. Yan, J. Alloys Compd. 851, 156772 (2021)
X. Li, A. Ito, Y. Sogo, X. Wang, R. LeGeros, Acta Biomater. 5, 508 (2009)
Acknowledgements
The authors acknowledge the financial support by the National Natural Science Foundation of China (No. 51961026).
Author information
Authors and Affiliations
Contributions
ZY: investigation, methodology, writing–originial draft. KF: data curation, writing–review & editing. SD: investigation, methodology, data curation, writing–review & editing. YC: methodology, data curation, writing–review & editing. HY: conceptualization, writing–review & editing. HS: resources, writing–review & editing. CL: software, resources, writing–review & editing. ZH: conceptualization, methodology, investigation, writing–review & editing, project administration.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Available online at http://springerlink.bibliotecabuap.elogim.com/journal/40195.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yu, Z., Feng, K., Deng, S. et al. Quasi-in-situ Observation and SKPFM Studies on Phosphate Protective Film and Surface Micro-Galvanic Corrosion in Biological Mg–3Zn–xNd Alloys. Acta Metall. Sin. (Engl. Lett.) 37, 648–664 (2024). https://doi.org/10.1007/s40195-023-01642-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40195-023-01642-4