
Abstract
Purpose of Review
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease for which there is no cure and treatments are at best palliative. Several genes have been linked to ALS, which highlight defects in multiple cellular processes including RNA processing, proteostasis, and metabolism. Clinical observations have identified glucose intolerance and dyslipidemia as key features of ALS; however, the causes of these metabolic alterations remain elusive.
Recent Findings
Recent studies reveal that motor neurons and muscle cells may undergo cell type-specific metabolic changes that lead to utilization of alternate fuels. For example, ALS patients’ muscles exhibit reduced glycolysis and increased reliance on fatty acids. In contrast, ALS motor neurons contain damaged mitochondria and exhibit impaired lipid beta oxidation, potentially leading to increased glycolysis as a compensatory mechanism.
Summary
These findings highlight the complexities of metabolic alterations in ALS and provide new opportunities for designing therapeutic strategies based on restoring cellular energetics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal, progressive, neuromuscular disorder affecting upper and lower motor neurons [1, 2]. The disease was first described in 1869 by the French neurologist Jean-Martin Charcot and received public attention in 1939, when the famous New York Yankees baseball player Lou Gehrig was diagnosed with ALS. A late-onset disease, ALS affects people between the ages of 40 and 70, with an average age of 55 at the time of diagnosis and 2–5-year survival time [1]. Exceptional cases of earlier disease onset or slow progressing have also been observed, with the most notable example being that of the renowned physicist Stephen Hawking who was diagnosed at the age of 21 and leads an incredibly productive life at 74. Based on US population studies, about 5600 people in the USA are diagnosed with ALS every year. According to the ALS CARE Database, 60% of the people with ALS in the database are men and 93% of patients in the database are Caucasian. Currently, there is no cure for ALS, and riluzole, the only approved drug, extends lifespan only by a few months [2].
ALS can be broadly categorized into two types: (1) familial (5–10% cases), an inherited form where there is at least one other known case of ALS in the family, and (2) sporadic (90–95% cases), which occurs seemingly at random, without any known family history. Superoxide dismutase 1 (SOD1) was the first gene to be implicated in ALS with several mutations identified as causative of the disease [3]. Several studies have shown that mutations in SOD1 lead to a toxic gain of function of the protein that alters proteostasis [4]. Since the discovery of SOD1 mutations in 1993, several loci have been implicated in ALS including VCP, TDP-43, FUS, ubiquilin 2, C9orf72, and profilin, among others; these and the molecular mechanisms they are linked to have been recently reviewed [5]. Of note, among these loci is TDP-43, which associates with pathological aggregates in 97% of ALS cases [6]. Given that, 2–3% of patients also harbor disease-causing mutations in its C-terminus domain; TDP-43 protein has emerged as a common denominator for the vast majority of ALS cases. In 2011, C9orf72 captured the attention of the field; expansions of non-coding G4C2 hexanucleotide repeats within C9orf72 represent the most common genetic cause of familial ALS affecting approximately one third of the patient population [7, 8].
ALS is a multifactorial disease accompanied by abnormalities in diverse cellular processes including RNA processing, proteostasis, and dysregulation of cellular energetics [9, 10]. Studies showing that ALS patients exhibit signs of hypermetabolism accompanied by dramatic weight loss indicate that cellular and/or systemic metabolic defects are involved in the onset and progression of the disease [10]. Indeed, studies in animal models indicate that defects in energetics precede denervation [10]; thus, metabolic dysregulation constitutes a potentially important therapeutic target. This notion is further supported by a recent phase 2 clinical trial that found high caloric diets to be safe and well tolerated in patients, with a significant reduction in adverse effects [11]. The purpose of this article is to review key research findings in the area of metabolic dysregulation in the pathophysiology of ALS and highlight the challenges and opportunities for developing therapeutic strategies aimed at restoring cellular energetics.
Systemic Metabolic Alterations in ALS
Clinical observations have shown that energy homeostasis is dysregulated in ALS [12, 13]. Hypermetabolism, defined as increased basal metabolic rate and energy expenditure, can be detected early in ALS and persists as the disease progresses [14]. Metabolic profiling using plasma from ALS patients also identified significantly altered metabolites consistent with alterations in aminoacid biosynthesis pathways, glucose metabolism, TCA cycle, and liver metabolism [12, 13]. Analyses of cerebral spinal fluid (CSF) from ALS patients found high levels of glucose and alpha-hydroxybutyrate, an early biomarker of insulin resistance [15]. As with patients, in the SOD1 mutant mouse, the most widely used animal model for ALS, metabolic abnormalities are not restricted to motor neurons, but also involve astrocytes and oligodendrocytes, and occur early on in disease [16].
Dietary Intervention
Characterizing systemic metabolic alterations in ALS provides opportunities for identifying risk factors and also for developing dietary intervention in patients. For example, a 2007 case-control study conducted in Japan showed that overall high intakes of carbohydrates and low intakes of fat, in particular saturated and monounsaturated fats, are associated with higher risk of developing ALS [17]. This supports the notion that a high-fat diet may be protective. Indeed, a recent study (European Prospective Investigation into Cancer and Nutrition (EPIC)) showed that increased prediagnosis body fat is associated with a decreased risk of death from ALS [18]. In addition, high levels of triglycerides and an increased LDL/HDL cholesterol ratio appear to increase survival in patients [19]. In contrast to these studies, a small double-blind, placebo-controlled, randomized phase 2 clinical trial found that while a high-calorie diet was protective, it was the high carbohydrate and not high-fat formulation that reduced adverse effects and increased patient survival [11]. While more and perhaps larger studies are needed to clarify these inconsistencies in clinical data, it seems that high-calorie diets are well tolerated and have therapeutic potential. These human studies are consistent with experiments in rodents showing that in mutant SOD1 mice, a high-calorie diet improves survival by 20%, while caloric restriction hastens disease onset, and animals tend to reach endpoint sooner compared to normally fed animals [20, 21]. Notably, it has recently been shown that feeding triheptanoin, the triglyceride of heptanoate, is a promising dietary intervention that provides an alternative fuel for improving oxidative phosphorylation and increasing ATP production in SOD1 mice [22]. Additional suggestions for dietary intervention aimed at restoring metabolic dysfunction by providing alternate fuels have been recently reviewed [23]. Although no dietary intervention has been reported to date in Drosophila models of ALS, hypermetabolism has also been observed in the context of TDP-43 overexpression in motor neurons, which led to a significant increase in pyruvate, the key metabolite linking glucose metabolism to the TCA cycle [24]. In the future, it will be interesting to see whether specific nutrient formulations are beneficial to specific patient populations depending on the underlying cause of disease and individual metabolic profiles.
Metabolic Syndrome and ALS
Insulin resistance and glucose intolerance, two key features of metabolic syndrome, have also been associated with ALS [10, 25]. Recently, Pradat and colleagues conducted a study with 21 sporadic ALS patients in the age range of 18–75 years and investigated glucose homeostasis after glucose load [26]. Briefly, patients were administered a dose of glucose, and blood glucose and plasma insulin levels were measured before and after the glucose bolus. Blood glucose and serum insulin levels were found to be significantly increased after glucose intake in ALS patients compared to controls. Additionally, fasting free fatty acid levels were significantly elevated in patients, linking impaired glucose tolerance and insulin resistance to impaired fatty acid metabolism, another feature of metabolic syndrome. In contrast, a recent report found no significant differences in total cholesterol, triglycerides, HLD, LDL, and LDL/HDL ratio between ALS patients and age and gender-matched controls [27]. In addition, Rafiq et al. found no significant link between serum lipid levels and survival in ALS [28].
Based on the fact that high levels of triglycerides, total cholesterol (i.e., dyslipidemia), and body mass index were found by several studies to be protective factors in ALS and given that diabetes mellitus (DM) type II is often associated with dyslipidemia and obesity, it was hypothesized that the presence of premorbid DM could be protective in ALS. Indeed, patients with DM had a significant later onset of ALS and a trend towards slower disease progression [29]. Interestingly, Pioglitazone (Pgz), a drug used to treat DM, improved locomotor function and increased survival in SOD1-G93A mice [30]. Pgz was independently identified as neuroprotective using a phenotyopic screen in Drosophila models of ALS based on TDP-43 and FUS; while Pgz improved locomotor function, it did not increase survival in flies [24]. Interestingly, a Pgz clinical trial was stopped due to lack of positive effects on survival [31]. In retrospect, when compared to the SOD1 mouse data, the fly model appears to be a more accurate predictor of Pgz’s effect in ALS patients. Taken together, these findings indicate the presence of systemic metabolic alterations in ALS, in both animal models (flies and mice) and human patients. It remains to be determined whether these defects are causative, correlative, or compensatory. We expect that animal models will provide these answers that are much needed to devise therapeutic strategies based on restoring metabolic function in ALS patients.
Carbohydrate Metabolism and ALS
Researchers have long sought metabolic molecular signatures specific to ALS patients. The identification of such signatures would provide clues to better understand disease pathology or may be used as a way to diagnose patients. As early as the 1960s, researchers identified changes in carbohydrate metabolism. A study of 10 ALS patients in the age range of 50–65 years, focused on conducting glucose tolerance tests and characterization of glycolytic products including lactic acid and pyruvic acid from venous blood, showed that a majority of the patients presented decreased blood glucose tolerance [32] consistent with several subsequent studies (see “Metabolic Syndrome and ALS” section). Interestingly, the same study [32] showed normal fasting figures or dynamics of the arteriovenous difference in glucose, inorganic phosphate, serum potassium, pyruvic acid, lactic acid, and serum aldolase levels in ALS patients compared to controls.
Additional evidence of altered carbohydrate metabolism comes from a study of 10 ALS patients from Guam, aged 40–63 [33]. After exposing study participants to intravenous arginine infusion, which stimulates insulin secretion, it was found that post infusion ALS patients showed a slight albeit not significant increase in glucose levels. The reduced glucose clearance together with findings of decreased insulin levels in serum of ALS patients compared to controls indicates impairments in insulin response and glucose metabolism as well as possible involvement of pancreatic islet cells in disease. Importantly, these observations were likely not attributable to muscle wasting in patients, since control subjects chosen in this study were age and weight-matched individuals also suffering from a muscular atrophy other than ALS.
To test the possibility that carbohydrate abnormalities result from a reduction in glucose transporter availability caused by muscle wasting, Reyes et al. measured in vivo insulin sensitivity in ALS patients and two control groups [25]. This study indicated that the insulin resistance in ALS could not be attributed simply to a reduction in muscle mass and availability of transporters but due to alterations in glucose metabolism associated with the disease process.
While several studies point towards glucose metabolic defects in ALS, contrasting results have also been reported. In a study published in 1984, Harno and colleagues were unable to demonstrate any marked decrease in glucose tolerance or increase in insulin resistance in ALS patients, compared to controls matched for age, obesity, and physical activity [34]. Later, Harris et al. were unable to detect defects in glucose or insulin sensitivity in ALS patients but found glucose disposal rates to be inversely related with disease severity [35]. Using PET scans, Ludolph and colleaues provided evidence for a moderate decrease of cerebral glucose metabolism in amyotrophic lateral sclerosis in parts of the brain other than the motor cortex [36]. These seemingly inconsistent results may be explained by the small sample size, making it difficult to generate a clear understanding of metabolic changes across the patient population.
It is possible that abnormalities in carbohydrate metabolism are specific to tissue type and may be missed in some cases when system-level measurements are performed. Indeed, Karpati and colleagues reported in chronically denervated forearm muscle of ALS patients an increased uptake of glucose and long-chain fatty acids from arterial blood accompanied by an abnormally high lactate concentration even in resting conditions [37]. Similarly Poulton and Rossi also observed increased glucose concentrations and enhanced glucose metabolism to fructose in skeletal muscle of ALS patients [38].
TDP-43 loss and gain of function mouse models exhibit changes in the expression of Tbc1d1, a Rab GAP involved in the translocation of perinuclear Glut4-containing vesicles in response to insulin [39, 40]. These results indicate that TDP-43 controls skeletal muscle glucose homeostasis.
In accordance with these observations, our lab has previously reported on the subtleties of tissue-specific interventions. Using a Drosophila model of ALS, we have shown that Pgz rescues neuronal and glial but not muscle-specific deficits induced by the overexpression of TDP-43 in these tissue types [24]. Pioglitazone activates peroxisome proliferator-activated receptor γ (PPARγ), a ligand-activated receptor that promotes the transcription of genes important in lipid and glucose metabolism. Our genetic interaction results coupled with drug treatment support the notion that PPARγ activation in the central nervous system but not muscles is partially protective in fly models of ALS.
Important gains in understanding ALS metabolism have also been made using in vitro approaches. Valbuena et al. employed NSC-34 cell lines, a spinal cord neuron-neuroblastoma hybrid, expressing either human wild type or mutant SOD1-G93A to identify metabolite changes [41•]. These experiments identified links between mitochondrial dysfunction, oxidative stress, and defects in ER stress response, all of which are implicated in ALS pathophysiology. Under serum deprivation conditions, the authors observed an increase in glycolytic protein expression and increased glycolytic flux to lactate in SOD1-expressing cells (both wild type and mutant). However, the net lactate flux was significantly higher in SOD1-G93A-expressing cells than in wild-type SOD1-expressing cells, an observation directly linked to decreased survival in the mutant cells. To explain this change in G93A-NSCs, the authors further showed that the protein expression levels of pyruvate dehydrogenase kinase 1 (PDK1) and lactate dehydrogenase A (LDHA, both enzymes critical for lactate overproduction) increased in these cells, while the glucose-derived acetyl-CoA flux into the TCA cycle decreased. In order to compensate for the increased glycolysis, the wild-type SOD1-NSCs increased the supply of amino acid for protein and glutathione synthesis, while this program failed in the G93A-NSCs leading to cytotoxicity. This work provides novel insights into the effects of impaired glucose metabolism causing poor stress adaptation and leading to neural damage in the presence of mutant SOD1. It will be important to confirm these changes in vivo and across multiple disease models.
Lipid Metabolism and ALS
Similar to alteration in carbohydrate metabolism, ALS patients also present with abnormal lipid profiles [19]. Conditional knockout of TDP-43 in mice resulted in accelerated fat loss in adipocytes through increased fat oxidation in these animals [39]. Notably, Tbc1d1, a gene involved in maintaining leanness, is significantly downregulated in the absence of TDP-43, thus linking TDP-43 to fat metabolism. Mutant SOD1 mice are leaner than wild-type littermates and exhibit characteristic hypermetabolism, mainly of muscular origin, as indicated by reduction in adipocity [21]. These defects can be detected in the asymptomatic stage, thus indicating that they are not only associated but may also contribute to neurodegeneration.
An overall increase in the use of peripheral lipid as energy source has also been observed. A recent study showed evidence of normal levels of plasma triglyceride and cholesterol under fasting conditions, but decreased levels of triglycerides, cholesterol, and total lipids under normal feeding conditions [42]. This leads to the question as to what might be the cause behind this alteration. Having excluded the possibility of decreased food intake, gastrointestinal malfunction, and misregulation of key factors involved in lipid biosynthesis in mSOD1 mice, the authors showed that lipids supplied by normal feeding rapidly disappear from the plasma of these animals, suggesting increased peripheral lipid clearance triggered by muscle hypermetabolism. This study lends further evidence to the “non-autonomous” involvement of muscle hypermetabolism in ALS disease progression.
Another study using asymptomatic SOD1G86R mice indicated decreased glucose handling in glycolytic muscles of the diseased animals [43•]. This is due to concerted effects caused by downregulation of the key glycolytic enzyme phosphofructokinase 1 (PFK 1) and upregulation of pyruvate dehydrogenase kinase 4 (PDK4), an enzyme that inhibits pyruvate dehydrogenase complex through phosphorylation, thus blocking conversion of pyruvate into acetyl-CoA. As a consequence, the lipid pathway was stimulated in these animals as early as during the presymptomatic stage and remained active till end stage of the disease, thus switching the fuel preference towards fatty acids by suppressing glucose utilization. Consistent with this, treatment with DCA, a specific inhibitor of PDK, restored normal messenger RNA (mRNA) expression of Pdk and Pfk1 mRNAs and resulted in decreased expression of denervation and atrophy markers. This was further translated at the functional level by restoration of muscle strength, larger muscle fibers, and overall weight gain in DCA-treated animals compared to non-treated SOD1G86R mice. Thus altogether, this and previous reports show that as the glycolytic muscles progressively lose their ability to utilize glucose, they switch to lipids as an alternate energy source and that this metabolic switch happens largely in the early presymptomatic stage.
Recent lipidomic analyses revealed significant alterations between muscle and spinal cord lipid profiles of presymptomatic and symptomatic SOD1 mice compared to wild-type littermates [44]. Further investigation revealed appreciable changes in sphingolipids, including ceramides and elevated levels of glulcosylceramides (GlcCer) and glycosphingolipids (GSLs) in muscles, as a result of upregulation of the GlcCer biosynthetic enzyme glucosylceramide synthase (GCS). On the contrary, inhibition of GCS in wild-type mice led to reversal of overexpression of many oxidative metabolism genes, indicating that modulation in the levels of GSLs may play a critical role in ALS muscle pathology and could be an interesting therapeutic target.
Mitochondrial Abnormalities
Mitochondrial dysfunction has been previously implicated in neurodegenerative diseases. An excellent overview of the mechanisms that lead to the accumulation of damaged mitochondria in ALS, particularly in SOD1 models, has been published recently [45]. Here, we highlight two novel mechanisms underlying defects in mitochondrial function that have recently been reported in TDP-43 and C9orf72 models of ALS and could explain the cellular energetic abnormalities in degenerating motor neurons. First, TDP-43 was reported to associate with the inner mitochondrial membrane via one or more import motifs [46••]. Mechanistically, TDP-43 associates with mitochondrial mRNA encoding the ND3 and ND6 subunits of the respiratory complex I and inhibits their translation leading to mitochondrial dysfunction and fragmentation. Using an elegant, peptide-based approach to inhibit TDP-43’s localization to mitochondria, the authors observe rescue of TDP-43-mediated toxicity in human cells. Second, dipeptide repeats, specifically (GR)80 produced by C9orf72 G4C2 expanded repeats, associate with mitochondrial ribosomes and lead to mitochondrial dysfunction and increased oxidative stress, which, in turn, causes DNA damage in induced pluripotent cell (iPSc) motor neurons [47••]. The mitochondrial abnormalities detected in motor neurons are expected to cause energy deficits that may lead to metabolic imbalance in ALS.
Conclusions
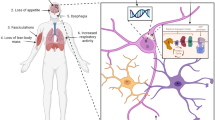
Years of clinical research have revealed systemic metabolic abnormalities in ALS patients. Although some inconsistencies persist, the majority of studies report some form of disturbance in functional metrics such as reduced glucose tolerance, insulin resistance, and abnormal utilization of fatty acids. These seemingly disparate findings are likely related to the small size of patient cohorts, which are limited by availability of eligible participants, and to the heterogeneous nature of patient populations with mixed etiologies and at different stages of disease. It is unarguably challenging to precisely determine in patients what physiological abnormalities are causative versus correlative, which, in turn, hampers the development of effective therapies. These challenges are countered by opportunities to pursue the mechanisms underlying metabolic dysregulation in animal models. These models provide the tools for genetic and dietary interventions that can distinguish between cause and consequence. For example, although the primary cause remains unknown, in ALS muscles, there seems to be a reduction in glycolysis that is compensated by utilization of alternate fuels such as fatty acids (Fig. 1). In contrast, in motor neurons, the impairment in mitochondrial function may lead to compensatory mechanisms that counterbalance the defects in oxidative phosphorylation such as increased glycolysis as reported in cultured cells or altered interactions with lactate-producing glial cells. In conclusion, our understanding of metabolic dysregulation in ALS is at a crossroads where challenges are turning into opportunities for elucidating the mechanisms underlying energy deficits in motor neurons, muscles, and glial cells. These advances in basic research together with the emerging power of personalized medicine are likely to lead to new insights and therapeutic strategies based on restoring cellular energetics in ALS.

Metabolic dysregulation in ALS. a Defects in mitochondrial function (see text for causes) lead to systemic metabolic changes and motor neuron degeneration. b Current data suggests that in ALS muscles, there is a compensatory upregulation of lipid utilization, while in motor neurons, glycolysis appears to be upregulated as a compensatory mechanism
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14(4):248–64. doi:10.1038/nrn3430.
Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012;124(3):339–52. doi:10.1007/s00401-012-1022-4.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi:10.1038/362059a0.
Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281(5384):1851–4.
Taylor JP, Brown Jr RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206. doi:10.1038/nature20413.
Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–38. doi:10.1016/j.neuron.2013.07.033.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56. doi:10.1016/j.neuron.2011.09.011.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68. doi:10.1016/j.neuron.2011.09.010.
Peters OM, Ghasemi M, Brown Jr RH. Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125(5):1767–79. doi:10.1172/JCI71601.
Dupuis L, Pradat PF, Ludolph AC, Loeffler JP. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011;10(1):75–82. doi:10.1016/S1474-4422(10)70224-6.
Wills AM, Hubbard J, Macklin EA, Glass J, Tandan R, Simpson EP, et al. Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2014;383(9934):2065–72. doi:10.1016/S0140-6736(14)60222-1.
Lawton KA, Cudkowicz ME, Brown MV, Alexander D, Caffrey R, Wulff JE, et al. Biochemical alterations associated with ALS. Amyotrophic lateral sclerosis : official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2012;13(1):110–8. doi:10.3109/17482968.2011.619197.
Lawton KA, Brown MV, Alexander D, Li Z, Wulff JE, Lawson R, et al. Plasma metabolomic biomarker panel to distinguish patients with amyotrophic lateral sclerosis from disease mimics. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2014;15(5–6):362–70. doi:10.3109/21678421.2014.908311.
Bouteloup C, Desport JC, Clavelou P, Guy N, Derumeaux-Burel H, Ferrier A, et al. Hypermetabolism in ALS patients: an early and persistent phenomenon. J Neurol. 2009;256(8):1236–42. doi:10.1007/s00415-009-5100-z.
Wuolikainen A, Jonsson P, Ahnlund M, Antti H, Marklund SL, Moritz T, et al. Multi-platform mass spectrometry analysis of the CSF and plasma metabolomes of rigorously matched amyotrophic lateral sclerosis, Parkinson’s disease and control subjects. Mol BioSyst. 2016;12(4):1287–98. doi:10.1039/c5mb00711a.
Sun S, Sun Y, Ling SC, Ferraiuolo L, McAlonis-Downes M, Zou Y, et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci U S A. 2015;112(50):E6993–7002. doi:10.1073/pnas.1520639112.
Okamoto K, Kihira T, Kondo T, Kobashi G, Washio M, Sasaki S, et al. Nutritional status and risk of amyotrophic lateral sclerosis in Japan. Amyotroph Lateral Scler. 2007;8(5):300–4. doi:10.1080/17482960701472249.
Gallo V, Wark PA, Jenab M, Pearce N, Brayne C, Vermeulen R, et al. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis: the EPIC cohort. Neurology. 2013;80(9):829–38. doi:10.1212/WNL.0b013e3182840689.
Dupuis L, Corcia P, Fergani A, Gonzalez De Aguilar JL, Bonnefont-Rousselot D, Bittar R, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. 2008;70(13):1004–9. doi:10.1212/01.wnl.0000285080.70324.27.
Hamadeh MJ, Rodriguez MC, Kaczor JJ, Tarnopolsky MA. Caloric restriction transiently improves motor performance but hastens clinical onset of disease in the Cu/Zn-superoxide dismutase mutant G93A mouse. Muscle Nerve. 2005;31(2):214–20. doi:10.1002/mus.20255.
Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci U S A. 2004;101(30):11159–64. doi:10.1073/pnas.0402026101.
Tefera TW, Wong Y, Barkl-Luke ME, Ngo ST, Thomas NK, McDonald TS, et al. Triheptanoin protects motor neurons and delays the onset of motor symptoms in a mouse model of amyotrophic lateral sclerosis. PLoS One. 2016;11(8):e0161816. doi:10.1371/journal.pone.0161816.
Tefera TW, Tan KN, McDonald TS, Borges K. Alternative fuels in epilepsy and amyotrophic lateral sclerosis. Neurochem Res. 2016; doi:10.1007/s11064-016-2106-7.
Joardar A, Menzl J, Podolsky TC, Manzo E, Estes PS, Ashford S, et al. PPAR gamma activation is neuroprotective in a Drosophila model of ALS based on TDP-43. Hum Mol Genet. 2014; doi:10.1093/hmg/ddu587.
Reyes ET, Perurena OH, Festoff BW, Jorgensen R, Moore WV. Insulin resistance in amyotrophic lateral sclerosis. J Neurol Sci. 1984;63(3):317–24.
Pradat PF, Bruneteau G, Gordon PH, Dupuis L, Bonnefont-Rousselot D, Simon D, et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11(1–2):166–71. doi:10.3109/17482960902822960.
Chio A, Calvo A, Ilardi A, Cavallo E, Moglia C, Mutani R, et al. Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology. 2009;73(20):1681–5. doi:10.1212/WNL.0b013e3181c1df1e.
Rafiq MK, Lee E, Bradburn M, McDermott CJ, Shaw PJ. Effect of lipid profile on prognosis in the patients with amyotrophic lateral sclerosis: insights from the olesoxime clinical trial. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2015;16(7–8):478–84. doi:10.3109/21678421.2015.1062517.
Jawaid A, Salamone AR, Strutt AM, Murthy SB, Wheaton M, McDowell EJ, et al. ALS disease onset may occur later in patients with pre-morbid diabetes mellitus. Eur J Neurol. 2010;17(5):733–9. doi:10.1111/j.1468-1331.2009.02923.x.
Schutz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schurmann B, et al. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25(34):7805–12. doi:10.1523/JNEUROSCI.2038-05.2005.
Dupuis L, Dengler R, Heneka MT, Meyer T, Zierz S, Kassubek J, et al. A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS One. 2012;7(6):e37885. doi:10.1371/journal.pone.0037885.
Ionasescu V, Luca N. Studies on carbohydrate metabolism in amyotrophic lateral sclerosis and hereditary proximal spinal muscular atrophy. Acta Neurol Scand. 1964;40:47–57.
Nagano Y, Tsubaki T, Chase TN. Endocrinologic regulation of carbohydrate metabolism. Amyotrophic lateral sclerosis and Parkinsonism-dementia on Guam. Arch Neurol. 1979;36(4):217–20.
Harno K, Rissanen A, Palo J. Glucose tolerance in amyotrophic lateral sclerosis. Acta Neurol Scand. 1984;70(6):451–5.
Harris MD, Davidson MB, Rosenberg CS. Insulin antagonism is not a primary abnormality of amyotrophic lateral sclerois but is related to disease severity. J Clin Endocrinol Metab. 1986;63(1):41–6. doi:10.1210/jcem-63-1-41.
Ludolph AC, Langen KJ, Regard M, Herzog H, Kemper B, Kuwert T, et al. Frontal lobe function in amyotrophic lateral sclerosis: a neuropsychologic and positron emission tomography study. Acta Neurol Scand. 1992;85(2):81–9.
Karpati G, Klassen G, Tanser P. The effects of partial chronic denervation on forearm metabolism. Can J Neurol Sci. 1979;6(2):105–12.
Poulton KR, Rossi ML. Peripheral nerve protein glycation and muscle fructolysis: evidence of abnormal carbohydrate metabolism in ALS. Funct Neurol. 1993;8(1):33–42.
Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, Wong PC. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A. 2010;107(37):16320–4. doi:10.1073/pnas.1002176107.
Stallings NR, Puttaparthi K, Dowling KJ, Luther CM, Burns DK, Davis K, et al. TDP-43, an ALS linked protein, regulates fat deposition and glucose homeostasis. PLoS One. 2013;8(8):e71793. doi:10.1371/journal.pone.0071793.
• Valbuena GN, Rizzardini M, Cimini S, Siskos AP, Bendotti C, Cantoni L, et al. Metabolomic analysis reveals increased aerobic glycolysis and amino acid deficit in a cellular model of amyotrophic lateral sclerosis. Mol Neurobiol. 2016;53(4):2222–40. doi:10.1007/s12035-015-9165-7. NSC-34 cell lines expressing a SOD1G93A variant show metabolic changes in stress response when deprived of serum. Specifically, G93A-NSCs show an increase in Pyruvate dehydrogenase kinase 1 (PDK1) and Lactate dehydrogenase A (LDHA), which promote lactate production.
Fergani A, Oudart H, Gonzalez De Aguilar JL, Fricker B, Rene F, Hocquette JF, et al. Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J Lipid Res. 2007;48(7):1571–80. doi:10.1194/jlr.M700017-JLR200.
• Palamiuc L, Schlagowski A, Ngo ST, Vernay A, Dirrig-Grosch S, Henriques A, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med. 2015;7(5):526–46. doi:10.15252/emmm.201404433. Pre-symptomatic SOD1G86R mice show reduced glucose processing and a switch to lipid dependency in glycolytic muscle.
Henriques A, Croixmarie V, Priestman DA, Rosenbohm A, Dirrig-Grosch S, D’Ambra E, et al. Amyotrophic lateral sclerosis and denervation alter sphingolipids and up-regulate glucosylceramide synthase. Hum Mol Genet. 2015;24(25):7390–405. doi:10.1093/hmg/ddv439.
Palomo GM, Manfredi G. Exploring new pathways of neurodegeneration in ALS: the role of mitochondria quality control. Brain Res. 2015;1607:36–46. doi:10.1016/j.brainres.2014.09.065.
•• Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016;22(8):869–78. doi:10.1038/nm.4130. TDP-43 localizes to the inner mitochondrial membrane where it inhibits the translation of mRNA crucial to respiratory complex I function.
•• Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, et al. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron. 2016;(2):92, 383–391. doi:10.1016/j.neuron.2016.09.015. GR(80) dipeptides produced by C9orf72 G4C2 expansion repeats cause mitochondrial dysfunction and increase oxidative stress in induced pluripotent stem cells.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Archi Joardar and Ernesto Manzo declare that they have no conflict of interest.
Daniela C. Zarnescu has a patent pending.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neurogenetics and Psychiatric Genetics
Rights and permissions
About this article

Cite this article
Joardar, A., Manzo, E. & Zarnescu, D.C. Metabolic Dysregulation in Amyotrophic Lateral Sclerosis: Challenges and Opportunities. Curr Genet Med Rep 5, 108–114 (2017). https://doi.org/10.1007/s40142-017-0123-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-017-0123-8