
Abstract
Purpose of Review
Knowledge of genetic etiologies for inherited cardiovascular disease has expanded in recent years giving providers the potential to tailor medical management and family screening based on a patient’s genotype for some conditions. This paper highlights recent advances in Mendelian inherited cardiovascular disease such as Marfan syndrome, hypertrophic cardiomyopathy, and familial hypercholesterolemia.
Recent Findings
Genetic testing has gone through a rapid evolution thanks to technological advancements in sequencing technology. The combination of increased utility and decreased cost is lowering the barriers for testing. Development of next-generation panels and whole-exome and whole-genome sequencing allows for broader testing and, in some cases, gene discoveries. Genetics is fueling growth in pharmacogenetics and complex disease research and has even led to novel therapeutics and new indications for old therapeutics.
Summary
The integration of clinical genetic testing into patient care is becoming increasingly common as the field expands and more institutions adopt the practice. The interpretation of genetic testing results can be complicated as there are a wide range of tools to determine the pathogenicity of a DNA variant identified in a patient. The psychological impact of genetic counseling necessitates pre- and post-test genetic counseling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease has been the leading cause of death in America every year since 1900 except 1918, the height of the great influenza pandemic. While most cardiovascular diseases remain unexplained (idiopathic), genetic etiologies to Mendelian cardiovascular diseases (CVDs) have been widely established, including hypertrophic cardiomyopathy (HCM) [1•], familial hypercholesterolemia (FH) [2•], arrhythmias such as long QT syndrome (LQTS) [3•], and aortopathies such as Marfan syndrome [4•]. When considered together, approximately 7.5/1000 individuals have the potential for a Mendelians form of CVD (Table 1). Like the examples above, many inherited CVD conditions have autosomal dominant inheritance patterns, meaning that all first-degree relatives of a proband have a 50 % chance of having a DNA variant that predisposes them to the condition.
Research into the molecular genetics of CVD has provided scientists and clinicians the background necessary to develop new therapeutics and treatment algorithms [5]. Understanding the genetic basis of disease has led to novel therapeutics (PCSK9 inhibitors for FH), novel applications for established drugs (losartan for Marfan syndrome), and targeted therapies for particular genetic defects (e.g., beta blockers vs sodium channel blockers for subtypes of LQTS) raising the hope of preventatively treating patients. Initiation of treatment before clinical manifestation (genotype positive, phenotype negative) has the potential to alter disease progression.
Importantly, the age of personalized medicine is rapidly dawning as epitomized by policy statements of the President of the United States thrusting genetics into the national spotlight [6]. Cardiovascular genetics is on the frontier of personalized (or precision) medicine. Advances in clinical genetic testing now allow refinement in diagnosis, management, treatment, and family screening based on genotype. Large-scale data sharing and reduced cost of genetic testing may facilitate unprecedented opportunities for clinical impact in patients with identified CVD. Educational institutions have responded by providing advanced degrees in genetic counseling, a field which has seen a 75 % increase in genetic counselors over the last decade. [7].
The current goals for clinical genetic testing are to aid diagnosis, inform medical management, and guide family screening. Conditions like Marfan syndrome, and LQTS, diagnosis, and/or treatment are partly guided based on the patient’s genotype. In the coming era of personalized medicine, the care team may tailor management and family screening based on a patient’s genotype. Advances in care, progress in clinical diagnostics, and attention to genetic counseling have significantly reduced the barriers to access for patients, most specifically reducing costs of screening and care [8, 9]. However, with increased opportunity comes increased responsibility to patients including variant interpretation, patient privacy, and informed consent during this data-driven era.
In this chapter, we first introduce the importance of pre- and post-test genetic counseling. Then we discuss the current use of genetics in cardiovascular clinical care, which is still mostly centered on the use and interpretation of limited gene panels. We then discuss how technological advances are allowing the use of high-throughput sequencing methods and highlight the role of genetic testing in diagnosing medical mysteries. We will briefly touch on an issue that will likely become more important over the next few years: the uses and limits of pharmacogenomics. Finally, we will provide a guide for using genetic testing in clinical applications and highlight cautionary tales from the field.
Clinical Significance of Genetic Counseling
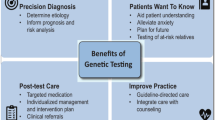
Genetic counseling is essential pre and post genetic testing [10•] (Fig. 1). During a pre-test visit, a genetic counselor will construct a detailed three-generation family history (pedigree) which will serve as the basis for family screening recommendations and yield subtle hints to disease in other family members such as syncope, or unexplained death in the family or relevant autopsy findings from relatives. Ultimately, the goal is to provide a risk assessment for the patient and their family. A genetic counselor will review benefits, limitations, and implications of genetic testing (such as insurance discrimination) to help the patient make an informed decision [11]. A solid psychosocial assessment, short-term patient-centered counseling, psychological support, and crisis interventions are some of the services genetic counselors can provide to patients and family members with an inherited CVD [12].

Incorporation of genetic testing in clinical care
Post-test genetic counseling allows for a better understanding of the genetic test results and family screening recommendations. Currently in the USA, physicians and genetic counselors are not able to directly contact family members due to HIPAA regulations. Therefore, a letter from a genetic counselor summarizing family screening recommendations and genetic testing options can serve as a remarkable tool for family members to discuss a diagnosis with each other and their local care providers [13].
Genetic counselors are well versed in the wide range of genetic testing options for patients with inherited CVD and assure the test with the highest yield is ordered. In an era of ever-expanding databases which aid in variant interpretation, genetic counselors will also help the providers and the patients make accurate conclusions from genetic testing.
From the administrative perspective, a consult with a genetic counselor is a billable encounter [14•]. Depending on the center, reimbursement can cover most of the costs of a genetic counselor. Given the positive impact of genetic counseling on patient care, clinics caring for inherited CVD should utilize a genetic counselor.
Genetic Basis Cardiovascular Diseases
The first DNA variants implicated in cardiovascular disease date to 1973 when Brown and Goldstein discovered that variants in LDLR cause FH [15]. Sanger sequencing was developed soon after which allowed for simple and rapid sequencing of single genes [16]. The mid-1980s and early 1990s saw the full dawning of cardiovascular genetics with the identification of genes known to cause HCM, LQTS, and Marfan syndrome. Advances over the last 25 years have resulted in multiple guidelines which now recommend the integration of genetic testing in clinical care (FH—[2•, 17••, 18•]; HCM—[19••]; LQTS—[20••]; Marfan—[21••]), and in 2013 major insurer Cigna adopted policies to insure genetic testing and to ensure that patients receive genetic counseling prior to testing [22]. A few examples will highlight the current roles of genetic testing in clinical settings and subtle differences that come from comparing and contrasting Marfan syndrome, HCM, and FH.
Marfan Syndrome and Aortopathies
The evolution of aortopathy genetic testing took a rapid and historic course. The discovery of the fibrillin-1 protein [23] was followed by immunohistochemical evidence of fibrillin-1’s role in patients with Marfan syndrome [24]. Specifically, linkage analysis has allowed localization of the FBN1 gene to 15q21.1 and further analysis identified multiple patients and families with defects in the FBN1 gene [25–27]. Within 5 years of its discovery, FBN1 genetic testing connected research to clinical application.
In the years since, our interpretation of DNA variants in FBN1 has changed dramatically. For example, cysteine substitutions in conserved cbEGF-like domains in the first 15 exons of the FBN1 gene often lead to more severe syndromic cases including ectopia lentis (lens dislocation) as a feature, and variants in exons 25–33 are more commonly associated with a more complex and severe clinical picture and younger onset. Marfan syndrome and variants that lead to loss of function of the fibrillin-1 protein often have less systemic involvement [28, 29]. Knowledge gained from protein domains, interactions, and function has also aided in variant interpretation. Recent papers have also suggested that the type of DNA variant may also affect how well a patient will respond to treatment [30•]. Approximately 85 % of patients with Marfan syndrome will have an identifiable DNA variant in FBN1.
Efficiencies in next-generation sequencing have fueled gene discovery. Genes have been associated with syndromic conditions (Loeys–Dietz syndrome) and non-syndromic familial thoracic aortic aneurysm and thoracic aortic dissection (FTAAD), which led to important distinction in disease course and medical management. For example, patients with ACTA2 DNA variants are more likely to present with acute dissection than patients with Marfan syndrome and one-third of individuals with ACTA2 variants dissected at dimensions smaller than 5.0 cm [31], which is the recommended surgical threshold for Marfan syndrome, but too liberal for patients with ACTA2 variants. These observations have propelled surgeons to modify the standard of care and customize surgical thresholds based on the patients’ underlying genetic etiology [4•]. Typically, surgical thresholds increase from the smallest to the largest aortic diameters based on variations in the following genes: SMAD3 = ACTA2 < TGFBR1/2 < FBN1 [21••]. The ACC’s 2010 guidelines for diagnosis and management of patients with FTAAD recognize that genetic testing provides the potential for early identification of individuals at risk. The guidelines recommend that patients with a family history of thoracic aortic aneurysms and dissections should undergo genetic testing. Approximately 20 % of patients with this history will have an identifiable DNA variant in one of a host of genes. Based on these results, first-degree relatives might further undergo genetic counseling and testing to determine who in the family is at risk and needs routine cardiovascular screening. Genetic testing is the standard of care for Marfan syndrome [21••].
Hypertrophic Cardiomyopathy (HCM)
Hypertrophic cardiomyopathy (HCM), an autosomal dominant genetic disorder of left ventricular hypertrophy, leading to an increased risk of sudden cardiac death and heart failure, is largely caused by DNA variants in sarcomere genes like MYBPC3 and MYH7 [32]. Previous estimates of 1/500 frequency of HCM may underestimate the true incidence which may be as high as 1/200 [1•].
Approximately 50 % of individuals with a diagnosis of HCM harbor an identifiable disease-causing DNA variant [19••] which can be identified using a multigene panel available from multiple labs. Identification of a disease-causing DNA variant allows family members to determine their relative risk for developing HCM and in some cases their risk for sudden cardiac death. Identifying family members at risk to develop HCM has allowed us to identify gene carriers who are at risk to develop HCM, but currently lack clinical signs or symptoms. This is especially important since evidence shows that gene carriers that have no clinical symptoms are still at increased risk [33]. Genetic testing for HCM is the standard of care and guidelines such as the 2011 ACCF/AHA guidelines for HCM recommend clinical screening of first-degree relatives and genetic testing in cases in which a DNA variant is identified in a proband [19••]. The last few years have shown a dramatic decrease in the out-of-pocket costs for genetic testing thanks to professional guidelines and clinical implications making it available at little or no cost to the patient (www.ncbi.nlm.nih.gov/gtr/).
As more probands are being genetically identified prior to symptom development, care providers are able to closely monitor the patient and detect subtle signs of the condition and treat early. Clinical trials are now targeting the so-called “genotype-positive, phenotype-negative” patients in the hopes of preventing the onset of symptoms. Currently, the VANISH clinical trial for patients with HCM is enrolling such patients in hopes the intervention will prevent the onset of HCM (ClinicalTrials.gov identifier: NCT01912534) [34]. If this is successful, predictive genetic testing for HCM would be given a whole new utility: the hope of prevention.
Familial Hypercholesterolemia (FH)
FH is an autosomal dominant genetic disease that leads to lifelong severe elevations in low-density lipoprotein cholesterol (LDL-C) and markedly increases the risk of coronary artery disease (CAD) versus that of the general population [2•]. FH has a rich history, including the Nobel Prize-winning work of Goldstein and Brown who identified the causal role of the LDL receptor protein in FH [35]. Later, pathogenic variants in the LDLR gene [15] and the APOB gene [36] were identified as causal for FH. More recently, the near-simultaneous observation that “activating” pathogenic variants in PCSK9 cause FH [37], while loss-of-function variants in PCSK9 cause low levels of LDL-C and CAD [38] led to the development of antibody-based PCSK9 inhibitors [39]. The marked LDL-C-lowering effect led to FDA approval of PCSK9 inhibitors for patients with FH and for non-FH patients with established CAD [40]. In addition, rare mutations in the LDLRAP1, ABCG5/ABCG8, and STAP1 genes have also been identified to be associated with autosomal recessive or dominant FH [41–43].
Similar to HCM, the classic estimates placed the prevalence at 1/500, but recent studies suggest that FH is as common as 1/200 and even more common in founder populations such as French Canadians and South African Jews [18•, 44–46, 94]. Thus, FH is the most common inherited CVD. When FH is identified and treated early enough with statin-based regimens, morbidity and mortality can be reduced by 76 % [47]. Unfortunately, fewer than 10 % of individuals with FH in the United States are diagnosed [18•]. Thus, FH is a prime example of a condition where enhanced screening efforts, especially cascade-based family screening, coupled with intervention could potentially save thousands of lives and millions of dollars. For this reason, the CDC has a Tier 1 recommendation for the use of genetic/genomic information (which may include genetic testing) in cascade screening efforts. Although not the case in the United States currently, genetic testing for FH is the standard of care in many European countries and evidence from several of these countries suggests that incorporating genetic testing into family-based cascade screening efforts improves the “pick-up” of diagnosis among family members [17••, 44, 48]. In fact, a nationwide opportunistic screening program based around genetic testing has proven successful in the Netherlands where a large fraction of the country’s FH population is diagnosed [18•]. Similar efforts on multiple continents have not only shown that this strategy has utility but is also cost effective [49, 50].
Generally, genetic testing for FH is currently based on 3 gene panels (Table 1) and involves sequencing all or part of LDLR, APOB, and PCSK9. In some countries where the spectrum of variants is limited, focused genotyping strategies have been employed. Genetic testing has not had deep penetration in the US market largely due to cost and access issues. However, as costs continue to decline this trend will likely change. Furthermore, randomized trials are underway to demonstrate the utility of genetic testing for improving cascade screening. In the “I FIGhT FH” trial [51], patients with likely FH are randomized to standard of care or standard of care plus genetic testing. The primary outcome is the number of relatives that undergo cascade screening.
Changing Landscape of Genetic Testing
In 1988, the National Institute of Health and the Department of Energy embarked on efforts to “coordinate research and technical activities related to the human genome.” The plan soon became commonly known as the Human Genome Project and was budgeted for $3 billion. At the turn of the millennium, scientists published the original drafts of the first genome sequence, ahead of time and under budget [52]. The ultimate cost was probably closer to $300 million as the genome project spurred technological advancement in genetic sequencing which increased throughput and lowered the price of sequencing.
Just a decade ago, sequencing a single gene costs thousands of dollars and had a turn-around time measured in months. The old Sanger sequencing technique was improved upon drastically, and soon multiple genes were able to be sequenced at once using next-generation techniques. Soon panels of multiple genes were available with an improved turn-around time and for the same cost as a single gene a decade ago. Current DNA sequencing panels range from 15 to 20 genes and often include a full analysis of deletion and duplication studies with cost for these panels being usually thousands of dollars; however, when indicated, most patients pay less than $100 for testing due to favorable financial assistance programs from genetic testing labs.
Next-generation sequencing now also allows sequencing of entire exomes (whole-exome sequencing, WES) or genomes (whole-genome sequencing, WGS) for ~$4000 and ~$8000, respectively. Research testing has been available for even cheaper. In a recent March 2016 announcement, Veritas became the first provider to offer clinical grade WGS for less than $1000, a long-touted goal. WES refers to sequencing the gene coding sequence of the genome, while WGS includes non-coding regions of the genome. This technological revolution is having profound changes in the clinical arena highlighted by the effect on “medical mysteries.”
As highlighted above, most of the current clinical genetic testing focuses on the interpretation of multiple gene panels. Just a few years ago, most of the sequencing itself was Sanger based and actually limited to a few genes and that is still sometimes the case. Increasingly, however, higher throughput approaches such as whole-exome sequencing are being utilized even if variant interpretation is limited to a circumspect list of candidate genes for the condition of interest. This is largely being driven by cost as it is now easier and cheaper to sequence an entire exome rather than a few genes. The tectonic shifts caused by advances in genetic sequencing technology cannot be understated.
Medical Mysteries
For a small subset of patients with rare conditions, the diagnostic odyssey can be frustrating, lengthy, and costly. In some cases, patients undergo years of standard clinical testing at multiple clinical sites to no avail. To alleviate this problem, in 2006 the NIH launched the undiagnosed disease program (UDP). With the decreased cost of sequencing, often WES or WGS is now included in part of the workup. Due to its initial success and oversubscription, the NIH’s Undiagnosed Disease Program expanded in 2015 and now includes seven regional sites which will encompass a much larger network and dramatically increase their bandwidth to 500 cases per year nationally [53•]. Individuals from around the country are able to submit cases for review through an online portal, and those that are accepted will receive a full battery of clinical testing at one of the centers which will include large-scale genetic testing to aid in diagnosis and discovery. During the pilot of the UDP, diagnoses were reached on 24 % of participants including three disorders diagnosed based off of single-nucleotide polymorphism array analysis and three others using whole-exome sequencing [54].
Instances of individual victories based on WGS have also become apparent recently. Physicians were able to complete full-genome sequencing on a newborn within days. It identified two variants, one of which causes long QT syndrome (LQTS). Pharmacotherapy was then tailored to his genotype and an ICD was placed before the newborn was discharged from the hospital [55].
Future Areas for Expansion
The prior examples have illustrated the current state-of-the-art in clinical genetic testing focused on Mendelian diseases. However, there have also been inroads in the areas of pharmacogenetics and in complex disease though, as the examples below will show, genetic testing in these areas is not being widely employed clinically in the US.
Pharmacogenetics
Genome-wide association studies (GWAS) have been used to identify SNPs that alter the way individuals metabolize medications including popular CVD medications such as clopidogrel, warfarin, and statins.
Three types of pharmacogenetic applications exist: (1) pharmacokinetic, (2) pharmacodynamic, and (3) underlying disease mechanism. Pharmacokinetic examples include warfarin sensitivity in which CYP2C9 variants decrease clearance by 30–90 % [56]. Pharmacodynamic examples include VKORC1 and CYP4F2 variants which result in an increased risk for an out-of-range INR value due to their dynamic effect on warfarin [57, 58]. An example of the final class of applications involves multiple haplotypes of APOE and their effect on statin response with the ε2 haplotype having greatest decrease when taking statins, followed by ε3 and ε4 [59]. Statin-induced myopathy is also linked to variants in SLC01B1 [60].
The application of pharmacogenetics has been hampered by a lack of adoption across the field. Although adoption has been slow, testing of this type has been made popular by direct-to-consumer companies which offer testing for many of these pharmacogenetic loci, and some WGS and WES providers offer patients the opportunity to receive these results. The Clinical Pharmacogenetics Implementation Consortium has recently provided a database of curated variants open to the public (pharmGkb.org).
Complex Disease: Benefit of Genetic Testing
Genetic discovery efforts, mostly GWAS, have now identified over 50 variants associated with CAD and hundreds associated with CAD risk factors such as hyperlipidemia [61], hypertension [62], and diabetes mellitus type 2 [63]. The common variants generally have low effect due to size with an odds ratio of <1.2 [64].
Efforts have been made to determine whether the use of genomic risk scores incorporating these variants predict CAD [65, 66]. And while these risk scores are predictive, the benefits above and beyond traditional risk scores (e.g., Framingham) are modest at best. Therefore, genetic testing for common and complex CVD has not been endorsed by guidelines in the absence of clinical trial data showing that this information increases outcomes. Such trials are underway [67] and these genetic risk score predictors may improve, especially as we learn more about the genetic architecture of CVD.
Benefits of Genetic Testing
Genetic testing has been shown to have many beneficial effects on patients. For instance, people may treat genetic information differently than other types of information, a concept known as “genetic exclusivity.” For instance, individuals that have undergone genetic testing for FH may be more adherent to medications leading to lower LDL-C levels [68], though this may reflect effects on provider behavior.
Additional benefits have been seen in families that have used genetic testing to aid in cascade screening. It has been shown to be cost effective in arrhythmia conditions [69] and to have a great diagnostic advantage as opposed to clinical screening alone in HCM [70]; this is also true for FH [17••, 18•].
Patients who undergo genetic testing of large scale (WGS, WES) have the additional option of receiving information about the American College of Medical Genetics (ACMG) incidental findings [71]. The incidental findings include a group of 56 genetic conditions that the AMCG determined are manageable and diagnosis, even secondarily, would have benefit on patients. This has been controversial due to the recommendation that these results should be disclosed to all pediatric and adult patients seeking testing. Studies have shown that approximately 3 % of WGS and WES results have reported incidental findings [72].
Access and Logistics of Genetic Testing
As prices decrease, the access to genetic testing is increasing and insurers are increasing access for their patients, especially given the cost effectiveness in family screening. Many labs now claim that 90 % of patients with commercial insurance pay less than $100 for guideline-based genetic testing out of pocket (GeneDx, Ambry Genetics). At this rate, clinical genetic testing may even hit a consumer price point in the next few years.
The favorable cost climate is helping ensure that more individuals receive potentially life-saving genetic testing. This has a twofold benefit. First, more individuals have access to genetic testing which was previously only available to a small few. Second, the more the people are genetically tested, the more we will be able to establish case data in an effort to more confidently apply criteria for variant interpretation. This has the overall effect of increasing the accuracy of testing outcomes, which in turn improves the quality and scope of personalized medicine.
Often, genetic testing companies take the burden of seeking insurance authorization on their own, helping simplify the process for providers. A test requisition, clinical information, family history, insurance information, and a small blood sample in a DNA-approved EDTA purple top tube are all that is needed to submit a patient sample. Including clinical information is essential in determining if the correct test is ordered and in aiding interpretation [73]. Turn-around time is typically a few weeks, up to a month. Once a variant is identified in a proband, family members may undergo single-site genetic testing while can be done with little DNA and enables testing in the prenatal realm or with very little tissue from a tumor sample or with saliva.
Caveats
Genetic testing has limitations. Given less-than-perfect yield, the absence of an identifiable pathogenic variant in a proband does not mean their condition is not genetic. Additional pitfalls that can lead to incorrect information are being disseminated to patients due to inconsistencies between labs’ interpretation of rare variation, and this is even occurring for well-established disease genes [74]. A series of cases presented to a specialty lab for second opinion revealed a 71 % discordance rate among their select cases [75]. Some genetic testing companies have access to private data (case data or functional data) that are not shared with the public. These instances lead to labs giving discordant recommendations, which has implications in patient care and family screening recommendations.
ClinGen was launched in 2013 to provide patients, clinicians, laboratories, and researchers a place to share genetic and phenotypic data. The goal is to provide a central resource that defines the clinical relevance of DNA variants for use in precision medicine and in research [76•]. A wide array of laboratories has already begun inputting data, and there is hope that this will help increase concordance among genetic testing laboratories.
In larger tests such as WES or WGS, these considerations can be magnified. Next-generation panel testing reads a single DNA base 100 times before determining what nucleotide fills that position [77]. However, not all regions of the genome and exome are accessible for sequencing under standard sequencing conditions. This results in areas of dropout in whole-genome and whole-exome testing in which a base is only read fewer than 20 times before making a call [77]. If a gene of interest had insufficient coverage, a panel test becomes necessary. Therefore, panel testing is currently the gold standard for many inherited CVDs because they are able to more confidently cover the full genes of interest with greater confidence due to specific reaction techniques in sequencing the genes [77]. This is why panels are used as a first-line test, and exome and genome as a second-line test in select cases. With so many options for providers, the utility of a genetic counselor is evident.
Federal protections have been put in place to protect patients from discrimination from a positive genetic test result thanks to the genetic information nondiscrimination act of 2008 (GINA) [78]. Protections include health insurance and employment discrimination, but fall short in supplemental insurances like disability and life insurance, and important point to mention when discussing the pros and cons of genetic testing with a patient.
Conclusion
Clincal genetic testing for CVD is standard of care in many inherited CVDs and clinically relevant in others. Novel tools are coming online to aid in the sharing of information and will help interpret genetic variation with more accuracy. Currently, genetic testing helps determine the appropriate medical management plan for many individuals with a variety of cardiovascular conditions ranging from cardiomyopathies to aortopathies. We are in a period of dynamic change and soon it will become a central pillar of diagnosis and treatment for single-gene disorders and holds promise for unlocking the secrets behind common “multifactorial” conditions.
In the next few decades, we will likely have treatments aimed at preventing the disease in pre-symptomatic gene carriers. President Obama’s promise of precision medicine is already at work in the cancer realm, where genetic tumor typing is the guiding treatment. Soon, similar advances will yield insight into battle against inherited CVD.
References
Recently published papers of particular interest have been highlighted as: • Of importance •• Of major importance
• Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249–54. Recent developments in detection such as genetic testing are shown to increase detection and in turn, the prevalence of hypertrophic cardiomyopathy from 1/500 to 1/200.
• Gidding SS, Champagne M, de Ferranti SD, Defesche J, Ito MK, Knowles JW, McCrindle B, Raal F, Rader D, Santos RD, Lopes-Virella M, Watts GF, Wierzbicki AS. The agenda for Familial Hypercholesterolemia, a scientific statement from the American Heart Association. Circulation. 2015;132(22):2167–92. Recent comprehensive statement from the AHA regarding the diagnosis, treatment and genetic testing and cascade screening options for patients and their families.
• Nakano Y, Shimogaki W. Genetics of long QT. J Hum Genet. 2016;61(1):51–55. Summarizes recent developments in Long QT genetic testing.
• Milewicz D, Hostetler E, Wallace S, Mellor-Crummey L, Gong L, Pannu H, Guo DC, Regalado E. Precision medical and surgical management for thoracic aortic aneurysms and acute aortic dissections based on the causative mutant gene. J Cardiovasc Surg. 2016;57(2):172–7. Current recommendations on surgical threshold for patients with aortic aneurysms and dissections.
O’Donnell CJ, Nabel E. Genomics of cardiovascular disease. New Engl J Med. 2011;365:2098–109.
The White House, O.O.T.P.S. FACT SHEET: president Obama’s precision medicine initiative. 2015.
Counselors, N.S.O.G. Professional status survey: salary & benefits. 2014: www.nsgc.org.
Ingles J, McGaughran J, Scuffham PA, et al. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart. 2012;98(8):625–30.
Wonderling D, Umans-Eckenhausen M, Marks D, Defesche JC, Kastelein JJ, Thorogood M. Cost-effectiveness analysis of the genetic screening program for familial hypercholesterolemia in The Netherlands. Semin Vasc Med. 2004;4(1):97–104.
• Hershberger RE, Siegfried J. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57:1641–49. Comprehensive review on familial dilated cardiomyopathy.
Tester DJ, Ackerman M. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation. 2011;123:1021–37.
Austin J, Semaka A, Hadjipavlou G. Conceptualizing genetic counseling as psychotherapy in the era of genomic medicine. J Genet Couns. 2014;23(6):903–9.
van der Roest WP, Roest WP. Family letters are an effective way to inform relatives about inherited cardiac disease. Am J Med Genet. 2009;149A:357–63.
• Harrison TA, Doyle D, McGowan C, Cohen L, Repass E, Pfau RB, Brown T, Billing for medical genetics and genetic counseling services. J Genet Couns. 2010;19(1):38–43. Comprehensive gude on how to get reimbursed for genetic counseling services.
Goldstein JL, Dana S, Brunschede GY, Brown MS. Genetic heterogeneity in familial hypercholesterolemia: evidence for two different mutations affecting functions of low-density lipoprotein receptor. Proc Nat Acad Sci. 1975;72(3):1092–6.
Sanger FC, Coulson R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94:441–8.
•• Excellence N.I.f.H.a.C. Familial hypercholesterolaemia: identification and management: NICE quality standard. 2013. Guidelines developed for FH by the https://www.nice.org.uk/guidance/qs41. Accessed 22 Mach 2016. British guidelines on familial hypercholesterolemia.
• Nordestgaard BG, Chapman M., Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Kees Hovingh G, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A, for the European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur Heart. 2013;34:3478–90. European guidelines for identifying and treating familial hypercholesterolemia.
•• Gersh BJ, Maron B, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary. J Thor Cardiovasc Surg. 2011;142(6):1303–38. United States guidelines on diagnosing and managing hypertrophic cardiomyopathy.
•• Priori Silvia G, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang C-E, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C, Ackerman M, Belhassen B, Mark Estes III NA, Fatkin D, Kalman J, Kaufman E, Kirchhof P, Schulze-Bahr E, Wolpert C, Vohra J, Refaat M, Etheridge SP, Campbell RM, Martin ET, Chye Quek S, HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2012;10(12):932–1963. HRS consensus statement on the diagnosis, treatment and management of arrhythmia syndromes.
•• Hiratzka LF, Bakris GL, Beckman JA, et al. ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: executive summary. J Am Coll Cardiol. 2010;55(14):509–1544. Guidelines on diagnosing and managing patients with thoracic aortic disease.
Office NE. Genetic counselling program gives Cigna customers increased access to genetic counselors. 2013.
Sakai LY, Keene D, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103(6):2499–24509.
Hollister DW, Godfrey M, Sakai LY, Pyeritz RE. Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. New Engl J Med. 1990;323(3):152–9.
Kainulainen K, Pulkkinen L, Savolainen A, Kaitila I, Peltonen L. Location on chromosome 15 of the gene defect causing Marfan syndrome. New Engl J Med. 1990;323:152–9.
Dietz H, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar E, Curristin S, Stetten G, Meyers DA, Francomano CA. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–9.
Dietz H, Saraiva J, Pyeritz RE, Cutting GR, Francomano CA. Clustering of fibrillin (FBN1) missense mutations in Marfan syndrome patients at cystein residues in EGF-like domains. Hum Mutat. 1992;1:366–74.
Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Marziliano N, Dietz HC, Halliday D, Beroud C, Bonithon-Kopp C, Claustres M, Muti C, Plauchu H, Robinson PN, Adès LC, Biggin A, Benetts B, Brett M, Holman KJ, De Backer J, Coucke P, Francke U, De Paepe A, Jondeau G, Boileau C. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81(3):454–66.
Faivre L, Collod-Beroud G, Callewaert B, Child A, Binquet C, Gautier E, et al. Clinical and mutation-type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24-32 mutation. Eur J Hum Genet. 2009;17:491–501.
• Franken R, Radonic T, Micha D, Maugeri A, van Dijk FS, Meijers-Heijboer HE, Timmermans J, Scholte AJ, van den Berg MP, Groenink M, Mulder BJ, Zwinderman AH, de Waard V, Pals G. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ Cardiovasc Genet. 2015;8(2):383–8. First finding that response to therapy in Marfan syndrome is based on genotype.
Milewicz D, Regalado E. Use of genetics for personalized management of heritable thoracic aortic disease: how do we get there? J Thoracic Cardiovasc Surg. 2015;149(2):S3–5.
Cirino AL, Carolyn Ho. Hypertrophic cardiomyopathy overview. Gene Reviews 2014; Available from: http://www.ncbi.nlm.nih.gov/books/NBK1768/.
Nannenberg EA, Michels M, Christiaans I, Majoor-Krakauer D, Hoedemaekers YM, van Tintelen JP, Lombardi MP, ten Cate FJ, Schinkel AF, Tijssen JG, van Langen IM, Wilde AA, Sijbrands EJ. Mortality risk of untreated myosin-binding protein C-related hypertrophic cardiomyopathy: insight into the natural history. Am Coll Cardiol. 2011;58(23):2406–16.
Valsartan for attenuating disease evolution in early sarcomeric HCM (VANISH). 2015. www.clinicaltrials.gov. Available from 22 03 2016.
Brown MS, Goldstein JL. Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc Natl Acad Sci USA. 1974;71(3):788–92.
Whitfield AJ, Barrett P, van Bockxmeer FM, Burnett JR. Lipid disorders and mutations in the APOB gene. Clin Chem. 2004;50:1725–32.
Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derré A, Villéger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–6.
Cariou B, Ouguerram K, Zaïr Y, Guerois R, Langhi C, Kourimate S, Benoit I, Le May C, Gayet C, Belabbas K, Dufernez F, Chétiveaux M, Tarugi P, Krempf M, Benlian P, Costet P. PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2009;29(12):2191–7.
Hooper AJ, Marais A, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193(2):445–8.
Garcia K. Federal panel backs approval of new drug to fight heart attacks. Routledge: New York Times; 2015.
Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, Calandra S, Bertolini S, Cossu F, Grishin N, Barnes R, Cohen JC, Hobbs HH. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292:1394–8.
Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–5.
Fouchier S, Dallinga-Thie GM, Meijers JCM, Zelcer N, Kastelein JJP, Defesche JC, Hovingh GK. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ Res. 2014;115(6):552–5.
Sjouke B, Kusters D, Kindt I, Besseling J, Defesche JC, Sijbrands EJ, Roeters van Lennep JE, Stalenhoef AF, Wiegman A, de Graaf J, Fouchier SW, Kastelein JJ, Hovingh GK. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart. 2015;36(9):560–5.
Benn M, Watts G, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J ClinEndocrinol Metab. 2012;97(11):3956–64.
Knowles JW, Youngblom E. Familial hypercholesterolemia. 2014. http://www.ncbi.nlm.nih.gov/books/NBK174884/. Accessed 21 March 2016.
Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DC, Liem AH, Witteman JC, Lansberg PJ, Kastelein JJ, Sijbrands EJ. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. Br Med J. 2008;337:a2423.
Hopkins P. Defining the challenges of familial hypercholesterolemia screening: introduction. J Clin Lipidol. 2010;4:342–5.
Mataa P, Alonsob R, Pérez-Jiménezc F. Screening for familial hypercholesterolemia: a model for preventive medicine. Rev Espanola Cardiol. 2014;67(9):685–8.
Ademi Z, Watts G, Pang J, Sijbrands EJ, van Bockxmeer FM, O’Leary P, Geelhoed E, Liew D. Cascade screening based on genetic testing is cost-effective: evidence for the implementation of models of care for familial hypercholesterolemia. J Clin Lipidol. 2014;8(4):390–400.
Rader D. Specialist perspective on FH risk and care continuum: stopping the progression from genetic mutation to vascular disease. In: FH 2014 Global Summit. 2014.
Consortium, I.H.G.S. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45.
• Gahl W, Wise AL, Ashely EA. The undiagnosed diseases network of the national institute of health. J Am Med Assoc. 2015;314(17):1797–98. Update on the current launch of the Undiagnosed Disease Program which is accepting applications from all physicians.
Gahl WA, Markello T, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, Wolfe L, Groden C, Godfrey R, Nehrebecky M, Wahl C, Landis DM, Yang S, Madeo A, Mullikin JC, Boerkoel CF, Tifft CJ, Adams D. The national institutes of health undiagnosed diseases program: insights into rare diseases. Genet Med. 2012;14(1):51–9.
Priest J, Scott R, Ceresnak MD, Frederick E, Dewey MD. Molecular diagnosis of long QT syndrome at 10 days of life by rapid whole genome sequencing. Heart Rhythm. 2014;11(10):1707–13.
Rettie AE, Korzekwa K, Kunze KL, Lawrence RF, Eddy AC, Aoyama T, Gelboin HV, Gonzalez FJ, Trager WF. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Chem Res Toxicol. 1994;5(1):54–9.
Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hörtnagel K, Pelz HJ, Lappegard K, Seifried E, Scharrer I, Tuddenham EG, Müller CR, Strom TM, Oldenburg J. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427(6974):537–41.
Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood. 2008;111:4106–12.
Voora D, Shah S, Reed CR, Zhai J, Crosslin DR, Messer C, Salisbury BA, Ginsburg GS. Pharmacogenetic predictors of statin-mediated low-density lipoprotein cholesterol reduction and dose response. Circ Cardiovasc Genet. 2008;1:100–6.
Ramsey LB, Johnson S, Caudle KE, Haidar CE, Voora D, Wilke RA, Maxwell WD, McLeod HL, Krauss RM, Roden DM, Feng Q, Cooper-DeHoff RM, Gong L, Klein TE, Wadelius M, Niemi M. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin Pharmacol Ther. 2014;96(4):423–8.
Global Lipids Genetics Consortium, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Do R, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkilä K, Hyppönen E, Isaacs A, Jackson AU, Johansson A, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikäinen LP, Magnusson PK, Mangino M, Mihailov E, Montasser ME, Müller-Nurasyid M, Nolte IM, O’Connell JR, Palmer CE, Perola M, Petersen AK, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridg RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney AS, Döring A, Elliott P, Epstein SE, Eyjolfsson GI, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJ, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimäki T, Lin SY, Lindström J, Loos RJ, Mach F, McArdle WL, Meisinger C, Mitchell BD, Müller G, Nagaraja R, Narisu N, Nieminen TV, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stancáková A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YD, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrières J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Järvelin MR, Jula A, Kähönen M, Kaprio J, Kesäniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, März W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njølstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PE, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BH, Ordovas JM, Boerwinkle E, Palmer CN, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Kathiresan S, Mohlke KL, Ingelsson E, Abecasis GR. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–83.
Parmar PG, Taal H, Timpson NJ, Thiering E, Lehtimäki T, Marinelli M, Lind PA, Howe LD, Verwoert G, Aalto V, Uitterlinden AG, Briollais L, Evans DM, Wright MJ, Newnham JP, Whitfield JB, Lyytikäinen LP, Rivadeneira F, Boomsma DI, Viikari J, Gillman MW, Stpourcain B, Hottenga JJ, Montgomery GW, Hofman A, Kähönen M, Martin NG, Tobin MD, Raitakari O, Vioque J, Jaddoe VW, Jarvelin MR, Beilin LJ, Heinrich J, van Duijn CM, Pennell CE, EArly genetics and lifecourse epidemiology consortium, Lawlor DA, Palmer LJ. International GWAS Consortium identifies novel loci associated with blood pressure in children and adolescents. Circ Cardiovasc Genet. 2016;9:807–18.
Morris AP, Voight B, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, Prokopenko I, Kang HM, Dina C, Esko T, Fraser RM, Kanoni S, Kumar A, Lagou V, Langenberg C, Luan J, Lindgren CM, Müller-Nurasyid M, Pechlivanis S, Rayner NW, Scott LJ, Wiltshire S, Yengo L, Kinnunen L, Rossin EJ, Raychaudhuri S, Johnson AD, Dimas AS, Loos RJ, Vedantam S, Chen H, Florez JC, Fox C, Liu CT, Rybin D, Couper DJ, Kao WH, Li M, Cornelis MC, Kraft P, Sun Q, van Dam RM, Stringham HM, Chines PS, Fischer K, Fontanillas P, Holmen OL, Hunt SE, Jackson AU, Kong A, Lawrence R, Meyer J, Perry JR, Platou CG, Potter S, Rehnberg E, Robertson N, Sivapalaratnam S, Stancáková A, Stirrups K, Thorleifsson G, Tikkanen E, Wood AR, Almgren P, Atalay M, Benediktsson R, Bonnycastle LL, Burtt N, Carey J, Charpentier G, Crenshaw AT, Doney AS, Dorkhan M, Edkins S, Emilsson V, Eury E, Forsen T, Gertow K, Gigante B, Grant GB, Groves CJ, Guiducci C, Herder C, Hreidarsson AB, Hui J, James A, Jonsson A, Rathmann W, Klopp N, Kravic J, Krjutskov K, Langford C, Leander K, Lindholm E, Lobbens S, Männistö S, Mirza G, Mühleisen TW, Musk B, Parkin M, Rallidis L, Saramies J, Sennblad B, Shah S, Sigurðsson G, Silveira A, Steinbach G, Thorand B, Trakalo J, Veglia F, Wennauer R, Winckler W, Zabaneh D, Campbell H, van Duijn C, Uitterlinden AG, Hofman A, Sijbrands E, Abecasis GR, Owen KR, Zeggini E, Trip MD, Forouhi NG, Syvänen AC, Eriksson JG, Peltonen L, Nöthen MM, Balkau B, Palmer CN, Lyssenko V, Tuomi T, Isomaa B, Hunter DJ, Qi L, Wellcome Trust Case Control Consortium, Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators, Genetic Investigation of ANthropometric Traits (GIANT) Consortium, Asian Genetic Epidemiology Network-Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Shuldiner AR, Roden M, Barroso I, Wilsgaard T, Beilby J, Hovingh K, Price JF, Wilson JF, Rauramaa R, Lakka TA, Lind L, Dedoussis G, Njølstad I, Pedersen NL, Khaw KT, Wareham NJ, Keinanen-Kiukaanniemi SM, Saaristo TE, Korpi-Hyövälti E, Saltevo J, Laakso M, Kuusisto J, Metspalu A, Collins FS, Mohlke KL, Bergman RN, Tuomilehto J, Boehm BO, Gieger C, Hveem K, Cauchi S, Froguel P, Baldassarre D, Tremoli E, Humphries SE, Saleheen D, Danesh J, Ingelsson E, Ripatti S, Salomaa V, Erbel R, Jöckel KH, Moebus S, Peters A, Illig T, de Faire U, Hamsten A, Morris AD, Donnelly PJ, Frayling TM, Hattersley AT, Boerwinkle E, Melander O, Kathiresan S, Nilsson PM, Deloukas P, Thorsteinsdottir U, Groop LC, Stefansson K, Hu F, Pankow JS, Dupuis J, Meigs JB, Altshuler D, Boehnke M, McCarthy MI, DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–90.
Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, Alver M, Armasu SM, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikäinen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, de Andrade M, de Vries PS, van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Huang J, Jalilzadeh S, Kessler T, König IR, Lannfelt L, Lieb W, Lind L, Lindgren CM, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FU, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, Stirrups KE, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples LA, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, Hall AS, Hamsten A, Harris TB, Hazen SL, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimäki T, Loos RJ, Melander O, Metspalu A, März W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O’Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ, Farrall M, CARDIoGRAMplusC4D Consortium. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121–30.
Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, Guiducci C, Perola M, Jula A, Sinisalo J, Lokki ML, Nieminen MS, Melander O, Salomaa V, Peltonen L, Kathiresan S. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376(9750):1393–400.
Goldstein BA, Knowles JW, Salfati E, Ioannidis JP, Assimes TL. Simple, standardized incorporation of genetic risk into non-genetic risk prediction tools for complex traits: coronary heart disease as an example. Front Genet. 2014;1(5):254.
Knowles J, Pavlovic A, McConnell MV, Ashley EA. A randomized trial of personal genomics for preventive cardiology: design and challenges. Circ Cardiovasc Genet. 2012;5(3):368–76.
Umans-Eckenhausen MA, Defesche J, van Dam MJ, Kastelein JJ. Long-term compliance with lipid-lowering medication after genetic screening for familial hypercholesterolemia. Arch Int Med. 2003;163(1):65–8.
Bai R, Napolitano C, Bloise R, Monteforte N, Priori SG. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circulation. 2009;2(1):6–15.
Havndrup O, Bundgaard H, Andersen PS, Larsen LA, Vuust J, Kjeldsen K, Christiansen M. Outcome of clinical versus genetic family screening in hypertrophic cardiomyopathy with focus on cardiac beta-myosin gene mutations. Cardiovasc Res. 2003;57(2):347–57.
Directors, A.B.O. ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet Med. 2015;17(1):68–9.
Yang Y, Muzny MD, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM. Molecular findings among patients referred for clinical whole-exome sequencing. J Am Med Assoc. 2014;312(18):1870–9.
Aronson SJ, Clark EH, Varugheese M, Baxter S, Babb LJ, Rehm HL. Communicating new knowledge on previously reported genetic variants. Genet Med. 2012;14(8):713–9.
Pariani MJ. Lessons learned from a re-review of FBN1 variants utilizing new tools in variant review. In: American college of medical genetics. Tampa: University of South Florida; 2016.
Pepin MG, Murray M, Bailey S, Leistritz-Kessler D, Schwarze U, Byers PH. The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genet Med. 2016;18(1):20–4.
• Rehm HL, Berg J, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, Plon SE, Ramos EM, Sherry ST, Watson MS. ClinGen, ClinGen—the clinical genome resource. New Engl J Med. 2015; 372(23): 2235–42. Update on the ClinGen project which is a public data sharing effort aid in interpretation of DNA variants.
Sun Y, Ruivenkamp C, Hoffer MJV, Vrijenhoek T, Kriek M, van Asperen CJ, den Dunnen JT, Santen GW. Next-generation diagnostics: gene panel, exome, or whole genome? Hum Mutat. 2015;36(6):648–55.
Hudson KL, Holohan MK, Collins FS. Keeping pace with the times—the Genetic Information Nondiscrimination Act of 2008. New Engl J Med. 2008;358(25):2661–3.
Van Driest SL, Ommen SR, Tajik AJ, Jamil Gersh BJ, Ackerman MJ. Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80(6):739–44.
Codd MB, Sugrue DD, Gersh BJ, Melton LJ 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy: a population-based study in Olmsted County, Minnesota, 1975–1984. Circulation. 1989;80:564–72.
Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–47.
Morales A, Hershberger R. Genetic evaluation of dilated cardiomyopathy. Curr Cardiol Rep. 2013;15(7):1–8.
Callis TE, Jensen BC, Weck KE, Willis MS. Evolving molecular diagnostics for familial cardiomyopathies: at the heart of it all. Expert Rev Mol Diagn. 2010;10(3):329–51.
Peters S. Advances in the diagnostic management of arrhythmogenic right ventricular dysplasia–cardiomyopathy. Int J Cardiol. 2006;113(1):4–11.
Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50(19):1813–21.
Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F, Nespoli L, Rimini A, Rosati E, Salice P, Spazzolini C. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120(18):1761–9.
Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, Bottelli G, Cerrone M, Leonardi S. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. J Am Med Assoc. 2005;294(23):2975–80.
Patel SS, Anees SS, Ferrick KJ. Prevalence of a Brugada pattern electrocardiogram in an urban population in the United States. Pacing Clin Electrophysiol. 2009;32(6):704–8.
Hedley PL, Jørgensen P, Schlamowitz S, Moolman-Smook J, Kanters JK, Corfield VA, Christiansen M. The genetic basis of Brugada syndrome: a mutation update. Hum Mut. 2009;30(9):1256–66.
Albornoz G, Coady MA, Roberts M, Davies RR, Tranquilli M, Rizzo JA, John A. Elefteriades, Familial thoracic aortic aneurysms and dissections—incidence, modes of inheritance, and phenotypic patterns. Ann Thoracic Surg. 2006;82(4):1405–6.
Dong SB, Zheng J, Ma WG, Chen MJ, Cheng LJ, He L, Xing QH, Sun LZ. Identification and surgical repair of familial thoracic aortic aneurysm and dissection caused by TGFBR1 mutation. Annal Vasc Surg. 2014;28(8):1909–12.
Rimoin D, Pyeritz RE, Korf B. Marfan syndrome and related disorders. In: Rimoin D, Pyeritz RE, Korf B, editors. Principles and practice of medical genetics. New York: Elsevier; 2013.
Loeys B, Backer JD, Van Acker P, Wettinck K, Pals G, Nuytinck L, Coucke P, De Paepe A. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mut. 2004;24(2):140–6.
Do R, Stitzie NO, Won HH, Jørgensen AB, Duga S, Angelica Merlini P, Kiezun A, Farrall M, Goel A, Zuk O, Guella I, Asselta R, Lange LA, Peloso GM, Auer PL, NHLBI Exome Sequencing Project, Girelli D, Martinelli N, Farlow DN, DePristo MA, Roberts R, Stewart AF, Saleheen D, Danesh J, Epstein SE, Sivapalaratnam S, Hovingh GK, Kastelein JJ, Samani NJ, Schunkert H, Erdmann J, Shah SH, Kraus WE, Davies R, Nikpay M, Johansen CT, Wang J, Hegele RA, Hechter E, Marz W, Kleber ME, Huang J, Johnson AD, Li M, Burke GL, Gross M, Liu Y, Assimes TL, Heiss G, Lange EM, Folsom AR, Taylor HA, Olivieri O, Hamsten A, Clarke R, Reilly DF, Yin W, Rivas MA, Donnelly P, Rossouw JE, Psaty BM, Herrington DM, Wilson JG, Rich SS, Bamshad MJ, Tracy RP, Cupples LA, Rader DJ, Reilly MP, Spertus JA, Cresci S, Hartiala J, Tang WH, Hazen SL, Allayee H, Reiner AP, Carlson CS, Kooperberg C, Jackson RD, Boerwinkle E, Lander ES, Schwartz SM, Siscovick DS, McPherson R, Tybjaerg-Hansen A, Abecasis GR, Watkins H, Nickerson DA, Ardissino D, Sunyaev SR, O’Donnell CJ, Altshuler D, Gabriel S, Kathiresan S. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102–6.
DeMott K, Nherera L, Shaw EJ, Minhas R, Humphries SE, Kathoria M, Ritchie G, Nunes V, Davies D, Lee P, McDowell I, Neil A, Qureshi N, Rowlands P, Seed M, Stracey H, Thorogood M, Watson M. Clinical guidelines and evidence review for familial hypercholesterolaemia: the identification and management of adults and children with familial hypercholesterolaemia. London: National Collaborating Centre for Primary Care and Royal College of General Practitioners; 2008.
Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG, McKnight D, Bai R, Suchy S, Friedman B, Tahiliani J, Pineda-Alvarez D, Richard G, Brandt T, Haverfield E, Chung WK, Bale S. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2015;369:1–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
Mitchel J. Pariani and Joshua W. Knowles declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Cardiovascular Genetics.
Rights and permissions
About this article

Cite this article
Pariani, M.J., Knowles, J.W. Integration of Clinical Genetic Testing in Cardiovascular Care. Curr Genet Med Rep 4, 107–118 (2016). https://doi.org/10.1007/s40142-016-0094-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-016-0094-1