
Abstract
Purpose of Review
Our goal is to provide a practical approach to the genetic evaluation of children with sensorineural hearing loss for use by practicing clinicians. We present the most recent research in the field followed by our recommended diagnostic algorithm incorporating genetic testing. We then provide case examples of commonly encountered patient presentations as further guidance.
Recent Findings
Genetic testing has the highest diagnostic yield of any single test in the evaluation of children with congenital bilateral severe-to-profound hearing loss. For unilateral or asymmetric hearing loss, imaging is typically more informative initially than genetic testing. Recent data show that syndromic forms of hearing loss are more common than previously appreciated.
Summary
A thorough and systematic evaluation of children with hearing loss that incorporates genetic testing leads to earlier diagnosis and treatment. This in turn leads to improved speech and language outcomes for children with hearing loss.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Permanent pediatric hearing loss is common, occurring in 1.7 in 1000 births (https://www.cdc.gov/ncbddd/hearingloss/data.html). This prevalence increases with age such that 31 in 1000 children have hearing loss by adolescence [1]. Newborn hearing screening (NBHS) has been implemented in the USA with the goal of early identification of affected children. In 2017, 3,742,608 children underwent NBHS, and 65,048 were subsequently identified to have hearing loss (https://www.cdc.gov/ncbddd/hearingloss/ehdi-data2017.html). After the initial failure on NBHS, an otolaryngologist is often the first point of contact for a patient and their family. Otolaryngologists should therefore be familiar with a framework for evaluation of these children with the goal of early diagnosis and treatment.
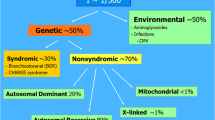
Hearing loss is classified as conductive, sensorineural, mixed (a combination of the two), and more rarely auditory neuropathy spectrum disorder (ANSD). Pediatric sensorineural hearing loss (SNHL) is, in the majority of cases, due to a genetic mutation. Additionally, and much less commonly, conductive hearing loss, mixed hearing loss, or ANSD may also have a genetic cause. These mutations may disrupt any portion of the incredibly complex molecular machinery responsible for the mechanotransduction of sound into an electrical stimulus. To date there are 7524 reported deafness-causing mutations in 120 non-syndromic hearing loss genes (Table 1, https://hereditaryhearingloss.org and http://deafnessvariationdatabase.org). In addition, there are several hundred known syndromes which include sensorineural hearing loss as part of the diagnosis. Non-genetic causes of childhood hearing loss include congenital infection, ototoxic medications, perinatal hyperbilirubinemia, or pre- or perinatal hypoxia, among others. Mitochondrial deafness is another form of genetic hearing loss which presents as progressive high-frequency loss due to mutations in mitochondrial genes that cause exquisite sensitivity to ototoxic medications [2].
Pediatric SNHL demonstrates marked clinical variability due to the myriad of possible causes. Hearing loss may range from mild to profound, it may affect one or both ears, and it may be progressive or stable. As described below, the clinical characteristics of hearing loss can guide evaluation. Conductive hearing loss is a very common cause of pediatric hearing loss and is most frequently caused by middle ear effusion or ossicular malformation. Some rare causes of genetic hearing loss may be associated with mixed or conductive hearing loss, for example, enlarged vestibular aqueduct or osteogenesis imperfecta. Diagnosis and treatment of conductive hearing loss is otherwise outside the realm of this review.
It is clear that children with untreated or ineffectively treated hearing loss have marked reduction in quality of life, speech and language development, and educational attainment [3, 4]. This holds true for unilateral hearing loss as well as mild-moderate hearing loss [5,6,7,8]. However, early diagnosis and treatment of childhood hearing loss with hearing aids or cochlear implants can lead to quality of life and educational attainment indistinguishable from peers without hearing loss [9,10,11].
The vast number of possible etiologies for childhood SNHL makes diagnosis difficult. However, accurate diagnosis of hearing loss is critical. There are four primary goals of evaluation of the child with SNHL: (1) to provide a unifying diagnosis, (2) to evaluate for syndromic causes of hearing loss and thereby identify comorbid disorders than may need to be addressed, (3) to provide prognostic information about hearing loss including progression and chance of occurrence in other family members, and (4) to guide further evaluation and treatment.
Over the past 10 years, advances in genetic testing have made it a cornerstone in evaluation of the child with hearing loss. In the next section, we will review these advances as well as some that are on the horizon.
Recent Advances in Genetic Testing for Deafness
The first genetic testing for SNHL emerged in the late 1990s after discovery of the GJB2 gene as a common genetic cause of hearing loss [12]. For the next decade, genetic testing was performed using either a panel of several common single mutations or single-gene sequencing. This testing was sequential and informed by clinical characteristics of the patient [13]. Given the extreme genetic heterogeneity of SNHL, these approaches had a relatively low diagnostic yield. For example, mutations in the most common deafness-causing gene, GJB2, were responsible for only 22.7% of diagnoses in a group of 1714 individuals with hearing loss and a genetic diagnosis [14]. The remainder of causative mutations were in one of the other 120 known deafness-causing genes. It was clear that an advance in the field of genetic testing for deafness was required to improve care of children with hearing loss.
Comprehensive Multi-Gene Panels
In 2010, new genetic sequencing techniques (termed massively parallel or next-generation sequencing) were used, for the first time, to perform sequencing of all known deafness genes simultaneously [15]. This type of testing, termed as comprehensive genetic testing or multi-gene panel testing, quickly became the gold standard in genetic evaluation of hearing loss [16]. Non-syndromic as well as syndromic causes of hearing loss are typically included, though the exact number of genes included is variable [17]. These platforms are now available from multiple laboratories and also differ based on the exact sequencing technique and analysis methods used but are routinely updated as new deafness-causing genes are discovered (Genetic Testing Registry, https://www.ncbi.nlm.nih.gov/gtr/).
Comprehensive genetic testing panels have improved the overall diagnostic yield of genetic testing for hearing loss. The two largest studies to date, encompassing more than a thousand subjects with hearing loss in each study who underwent testing using these panels, showed a remarkably similar diagnostic rate of about 40% [18, 19]. Similarly, a review that incorporated data from twenty studies with a total 603 patients of all ages with variable severity of hearing impairment showed an overall diagnostic rate of 41% [16].
Comprehensive genetic testing panels have therefore greatly improved the ability to diagnose genetic causes of hearing loss in children. It is important to note that the diagnostic rate of these comprehensive panels varies by clinical features such as family history, ethnicity, severity and symmetry of hearing loss, and syndromic features found on physical examination [18]. For instance, in cases of asymmetric and unilateral hearing loss, an anatomic abnormality is more likely to be the cause. One study showed a diagnostic rate of finding a genetic cause in 44, 22, and 1% of patients with bilateral symmetric, asymmetric, and unilateral hearing loss [18]. A subsequent study confirmed these results, with a genetic cause identified in 23% of children with unilateral and asymmetric hearing loss, while imaging revealed a significant finding in 46% of these children [20]. For children with congenital hearing loss, the diagnostic rate is approximately 53%, compared with 29% for children with later-onset hearing loss [14]. These differences in genetic diagnostic rates should be considered when ordering tests. Regardless, comprehensive genetic testing has emerged as the test with the single highest diagnostic rate in evaluation of children with SNHL.
Syndromic Hearing Loss
Comprehensive genetic testing panels typically evaluate for non-syndromic as well as syndromic causes of hearing loss. This is important given that many children with syndromic forms of hearing loss may not initially demonstrate clinically apparent syndromic findings on examination. For this reason, the term non-syndromic hearing loss “mimics” has been coined (Table 1). For example, children with Usher syndrome type I, the most severe form, have clinically apparent delayed motor milestones at 1–2 years of age and vision difficulties manifesting as night blindness in their pre-teen years. Similarly, children with Pendred syndrome will typically not have evidence of thyroid dysfunction until teenage years, and those affected by Jervell and Lange-Nielsen syndrome may not demonstrate cardiac symptoms attributable to long QT syndrome until sudden syncope or death [21]. Diagnosis of these syndromic forms of hearing loss is a central goal of evaluation of children with hearing loss, and comprehensive multi-gene panels are the best method to make this diagnosis.
The high prevalence of non-syndromic “mimics” was not understood until recently. In one cohort of 1119 adults and children with hearing loss who underwent comprehensive genetic testing, 23% of those with a genetic diagnosis were found to have a syndromic form of hearing loss [18]. The most common causes were Usher syndrome and Pendred syndrome followed by deafness-infertility syndrome. Although more studies are needed to replicate these findings, a positive test for one of these “mimics” means that additional medical evaluation will be needed.
Exome and Genome Sequencing
Exome sequencing has proved hugely influential in the diagnosis of rare genetic diseases and has emerged as a viable alternative to panel based testing for genetic hearing loss [22,23,24]. Exome sequencing refers to the process of sequencing every exon of every coding gene in the genome. By sequencing all genes in the genome, analysis can be performed not just on deafness-causing genes but also on genes with mutations in genes not previously associated with hearing loss. Further, as additional deafness-causing genes, family history and phenotype information are discovered the same DNA sample, and exome sequencing data can be used for re-analysis with a chance of identifying a definitive cause for the hearing loss [25].
However, by sequencing all genes, there are risks to discovering genetic mutations of uncertain clinical significance (VUS) or in genes of potential clinical importance but not related to hearing loss when exome sequencing is performed. In addition, because all genes are sequenced, sequencing coverage of hearing loss genes may be lower than multi-gene panels which focus solely on the hearing loss genes [22]. Other disadvantages include the cost to store and analyze the data, which is greater than targeted panels due to the increased number of genes sequenced. For any next-generation sequencing test, the method used for bioinformatics data analysis is critical and varies significantly between laboratories as each panel is developed in house. In contrast, exome sequencing is a widely used and more standardized tool. This makes the analysis and interpretation of exome data relatively more straightforward due to development of analytic best practices [26].
Genome sequencing expands upon exome sequencing to include every base pair of the human genome, totaling 3.2 billion bases. Whereas exome sequencing includes only coding regions of the genome, genome sequencing includes evaluation of non-coding and inter-genic regions which may be crucial for gene function and regulation. Genome sequencing has proven to be advantageous for other genetic diseases but has not yet been routinely integrated for testing in patients with hearing loss [27]. The primary advantage of genome sequencing is that the entire genome is sequenced so there is no concern for missed base pairs. However, the disadvantages of the cumbersome analysis and off-target results of exome sequencing are even greater for genome sequencing because of the additional genetic information obtained. The clinical interpretation of non-exonic genomic variants is often uncertain. In addition, there are greatly increased genetic counseling needs as more of the genome is sequenced. As sequencing costs continue to fall, it is foreseeable that genome sequencing will become the most standardized approach to uncovering a genetic cause of hearing loss. For now, comprehensive multi-gene panels and exome sequencing are the more practical options.
Genetic Newborn Hearing Screening
Recently, the addition of some form of genetic testing has been proposed as a way to improve the NBHS [14]. It is hypothesized that incorporating genetic screening would improve sensitivity, decrease time to diagnosis, and reduce loss to follow up for the current, physiologic-only, NBHS. Proposed approaches range from screening of several mutations in only the most common deafness genes to exome or genome sequencing. Several recent studies have examined the use of universal screening of all children for a handful of common deafness-causing mutations and found improved sensitivity of the NBHS overall in that more children with hearing loss were ultimately identified [28,29,30]. These platforms are cost-effective but work best in a homogeneous ethnic population due to the variability of deafness-causing mutations by ethnicity [14, 31]. Exome sequencing, while more expensive, could also play a role in NBHS. In one study, exome sequencing had a 56% diagnostic yield for infants who failed their physiologic NBHS and were found to have bilateral moderate to profound SNHL [24]. In this study, exome sequencing was integrated as a test after failure of NBHS and not as a universal screening approach. At the current time, integration of genetics into the NBHS is not cost-effective but may be in the near future as sequencing costs continue to fall.
Practical Approach to Evaluation of the Child with Hearing Loss
Over the past decade, genetic testing has become essential in the evaluation of children with hearing loss [32, 33]. However, no form of testing replaces careful clinical evaluation and expert judgment. As we will detail below, the initial clinical evaluation guides subsequent workup which varies significantly by age and clinical presentation.
Broadly speaking, there are four categories of childhood sensorineural hearing loss: (1) non-syndromic genetic hearing loss, (2) syndromic genetic hearing loss which may present as non-syndromic (non-syndromic “mimic”), (3) congenital infections, of which congenital cytomegalovirus is the most common, and (4) anatomic abnormalities of the cochleovestibular apparatus and the auditory nerve (many of which have a genetic basis). This is an oversimplification of a complex topic but can provide a broad differential diagnosis and thus framework for practical evaluation of children with hearing loss (Table 2).
For newborns with bilateral severe to profound hearing loss, genetic causes (either non-syndromic or syndromic) are highest on the differential. This is followed by environmental causes of hearing loss, in particular congenital cytomegalovirus, followed by anatomic abnormalities. For children with asymmetric hearing loss, congenital cytomegalovirus and anatomic abnormalities are more likely. If the hearing loss is mild-moderate and progressive but symmetric, genetic hearing loss is highest on the differential followed by anatomic abnormalities. Box 1.
Clinical evaluation of a child with hearing loss should begin with a careful history. Details of the pregnancy, birth history, and past medical history are crucial. Of specific concern are maternal infections and any stay in in the neonatal intensive care unit which predispose to non-genetic causes of hearing loss. Family history should include three generations, age of onset of hearing loss, and specifically include any form of hearing loss.
Physical examination should include a complete head and neck examination with special attention to possible syndromic features. White forelock, ear pits or tags, branchial cleft anomalies, cleft lip and palate, synophrys, vitiligo, café-au-lait spots and other abnormalities may guide diagnosis. Evaluation should include consideration of conductive or mixed causes of hearing loss, specifically otitis media or ossicular chain abnormalities. However, some forms of syndromic hearing loss like branchio-oto-renal (BOR) syndrome may present as a purely conductive hearing loss, and these should be considered.
Audiometric evaluation should then be performed using age-appropriate measures. Diagnostic auditory brainstem response and otoacoustic emissions are typically the first tests used in infants who fail NBHS. After infancy, developmentally appropriate audiometric testing may include behavioral audiometry, play audiometry, visual reinforcement audiometry, or pure tone testing. At any age, impedance audiometry, otoacoustic emissions, and acoustic reflex testing can also be performed to obtain further information.
Genetic Testing
Genetic testing is the most important test for diagnosis of bilateral congenital and childhood hearing loss. It has a lower, but still significant, diagnostic yield in children with asymmetric and unilateral hearing loss. In these children, imaging has the highest diagnostic yield. As described in the section above, genetic testing will typically take the form of multi-gene panels or exome sequencing with a focus on known deafness genes. As detailed above, the composition of these panels or the deafness genes evaluated during exome sequencing analysis varies significantly by laboratory.
The clinician ordering the test should have an understanding of the type and complexity of the genetic test being ordered and be able to provide pre- and post-test counseling to the child’s family after results of the test are available. This will often necessitate consultation with a geneticist or a genetic counselor. The turnaround time for these tests is typically on the order of 1–3 months. In some instances, single gene testing or limited panels may be the only available test or required prior to ordering full panels due to insurance coverage (see section below). In this case, testing of GJB2/6 (connexin 26/30) still has the highest yield, though in some ethnic populations, the yield of connexin testing is essentially zero [31]..
There has been a growing acceptance of the importance of genetic testing in the evaluation of children with hearing loss [16, 34]. Genetic testing may decrease the overall number of other diagnostic studies ordered and therefore total number of tests [34]. Despite this, in some cases, genetic testing for childhood hearing loss is still not recognized as medical necessity by some insurance companies. Pre-authorization should be attempted prior to ordering, as these tests typically cost several thousand dollars. If only a single gene or small panel is authorized, this should be performed first, and, if negative, further broader genetic testing should be pursued instead of declaring the child to not have genetic hearing loss. Additionally, if a particular syndromic diagnosis is suspected, or if there is an established genetic diagnosis in another family member, testing limited to those genes/mutations can be considered as an initial study.
Congenital Cytomegalovirus Testing
Congenital cytomegalovirus (cCMV) infection is the most common non-genetic cause of SNHL in newborns, occurring in 0.5–2% of all live born infants [33, 34, 35]. cCMV typically causes hearing loss that is characterized by its variability. It is frequently, but not always, progressive, asymmetric, and congenital. Diagnosis of cCMV requires an early suspicion to allow for testing prior to 3 weeks (21 days) of age. Testing in children greater than 3 weeks of age old is less accurate as the incidence of postnatally acquired CMV rises. If a child outside the newborn period is suspected of having cCMV, testing of dried blood spots from a newborn screening card may be able to establish the congenital nature of infection. Reported success of this varies based on the DNA extraction method used and the very small volume of usable DNA, with high specificity but variable sensitivity [35]. Hearing loss due to cCMV is potentially medically treatable with antivirals in the newborn period, and so early diagnosis is paramount [36,37,38]. Testing varies by laboratory but most often is a viral culture or PCR from saliva and urine. Universal cCMV screening has been advocated by several groups, but currently only 5 states, CT, IA, NY, UT, and VA, have mandated cCMV testing for all infants who fail the NBHS (ref 35 and https://www.nationalcmv.org).
Newborns with any form of SNHL should be tested for cCMV. Testing in older children is typically reserved for those with progressive, asymmetric hearing loss unexplained by imaging findings or other workup. In a recent study, 26% of children with unexplained SNHL were found to have cCMV based on positive DBS testing [39].
Importantly, a diagnosis of cCMV hearing loss does not preclude a diagnosis of genetic hearing loss and vice versa. In other words, these two etiologic causes of hearing loss may exist simultaneously in the same child. This is demonstrated in a recent study which documented this co-occurrence and estimated that about 2% of children with cCMV will also have a genetic cause of hearing loss [40] This finding has implications for treatment of cCMV hearing loss. Genetic testing for children with cCMV hearing loss should be based on severity and type of hearing loss. Typically, children with cCMV hearing loss will have asymmetric and progressive hearing loss. A child presenting with bilateral severe to profound hearing loss who tests positive for cCMV, for example, could be considered to be affected by a co-existent genetic cause of hearing loss and should be tested for such.
Imaging
Imaging for childhood hearing loss is crucial to guide diagnosis and treatment [20, 41, 42]. The timeframe and best method for imaging has been debated [43]. High-resolution computed tomography (CT) of the temporal bones provides anatomical and surgical detail that is familiar to the otolaryngologist. Due to the advent of ultrafast scanners, CT can now typically be performed without sedation. However, CT is associated with radiation which may lead to later development of malignancy [43]. In addition, CT does not completely evaluate the cochlear nerve, the absence of which remains the major anatomical contraindication to cochlear implantation.
MRI of the temporal bones provides fine-scale resolution of the cochlear nerve and can differentiate most cochleovestibular anomalies. However, given the longer time to obtain the imaging, sedation is often required. Diagnostic rates are similar between CT and MRI but with somewhat different findings and incidental findings [41, 42]. Many centers initially recommend MRI, typically first attempted in infants using a “feed and swaddle” technique. If this is unsuccessful, then sedation may be used. However, surgical planning prior to cochlear implantation may necessitate a temporal bone CT at a later date, and parents should be counseled on this.
Other Testing and Referrals
Children presenting with bilateral SNHL should have an electrocardiogram to evaluate for long QT syndrome (Jervell and Lange-Nielsen syndrome, JLN). This is a very rare autosomal recessive syndrome which may present with syncope or sudden death; identification is crucial for short- and long-term medical management of the cardiac defect, including safe anesthesia [21].
Referral to an ophthalmologist is similarly important. Decreased sensory input from hearing loss should not be compounded by an unnoticed secondary sensory deficit of vision loss or other ocular abnormalities. Early ophthalmologic evaluation and treatment of visual loss will provide best outcomes for children with hearing loss. In older children, retinal abnormalities may suggest a diagnosis of Usher syndrome or other syndromes involving retinal degeneration and hearing loss; these however may be difficult to detect in infants and young children, so a “normal” routine ophthalmologic evaluation does not always rule out genetic retinal dystrophies.
Case Examples
These following cases are presented to provide practical guidance in the face of a difficult diagnostic task. They are not meant to serve as “recipes” and clinical judgment is paramount.
Case 1: A Newborn with Severe-to-Profound Bilateral Sensorineural Hearing Loss
The patient is a 12-day-old female born at 38 5/7 to a G2P2 mother. The pregnancy was uncomplicated. Delivery was via a planned C-section and was atraumatic with APGARs of 8 at 1 min and 10 at 5 min. The child spent no time in the NICU and was discharged on day of life 2. The inpatient NBHS (AABR) showed refer bilaterally. Family history is significant for an older brother, 3 years old, who is healthy and passed his NBHS. Mother’s father wears hearing aids, otherwise no family history of hearing loss. Audiometry via a diagnostic ABR today performed prior to the visit under natural sleep showed severe-to-profound SNHL bilaterally with no responses at > 90 dB. On the exam, the patient has no cleft lip/palate, no ear pits/tags, and no syndromic facial features. External auditory canals are clear bilaterally with no middle ear fluid.
Broad differential diagnosis (in order of likelihood): Non-syndromic genetic hearing loss, syndromic genetic hearing loss (non-syndromic “mimic”), cCMV and other infections, and anatomic cochleovestibular anomaly including nerve deficiency.
Next Steps
-
Treatment: The Joint Committee on Infant Hearing (JCIH) 1-3-6 paradigm should be followed [44]. The patient has now received screening before 1 month, diagnosis before 3 months, and so is on track. The child should be fitted with hearing aids for a trial prior to 6 months but ideally as soon as possible. Ongoing discussion with the parents should focus on a likely need for cochlear implant.
-
Testing: Genetic testing should be performed, ideally using a multi-gene comprehensive panel or exome sequencing. In this child, there is a 50–60% chance of obtaining a diagnosis based on genetic testing alone. The patient is still within the 3-week window of cCMV testing, and so this should be performed. EKG should be ordered and referral to ophthalmology for evaluation of a possibly second sensory dysfunction.
-
Imaging: Imaging could be deferred pending the genetic testing results; however, given the likelihood of a cochlear implant for this child, an MRI is indicated. MRI should be performed, while the patient can still be fed and swaddled, typically < 3–6 months to prevent the need for sedation.
Case 2: A Young Child with Asymmetric Sensorineural Hearing Loss
The patient is a 2-year-old male with progressive, asymmetric sensorineural hearing loss. He failed NBHS on the right. He wears a hearing aid on the right. He recently started having worsening hearing on the left. He has slightly delayed speech, but otherwise is healthy with normal motor milestones and development. Brother (6 years old) is healthy. He is currently in kindergarten. No family history of hearing loss. Examination is within normal limits with no syndromic findings.
Differential diagnosis (in order of likelihood): cCMV and other infections, anatomic cochleovestibular anomaly including nerve deficiency, syndromic genetic hearing loss, and non-syndromic genetic hearing loss.
Next Steps
-
Treatment: Continue hearing aid use on the right and consider hearing aid use on the left. Follow audiograms closely particularly given recent change.
-
Testing: For this asymmetric and progressive hearing loss, testing should initially focus on non-genetic causes of hearing loss. Antibody testing for cCMV may provide some insight but is not specific. If it suggests prior CMV infection, the testing of the dried blood spot cards if available could be tested for cCMV. Genetic testing has a lower diagnostic rate for children with asymmetric hearing loss.
-
Imaging should be performed and based on discussion with the parents that would include a sedated MRI versus a temporal bone CT.
Conclusions
Early diagnosis and treatment are key to the best outcomes for children with hearing loss. Otolaryngologists are on the front line in caring for these patients. Genetic testing, specifically in the form of comprehensive multi-gene panels and exome sequencing, is critical in evaluation of children with hearing loss. Genetic testing guides further evaluation and treatment including providing prognostic information, allows for evaluation of syndromic forms of hearing loss, and provides recurrence risk information for parents.
Given the rapid recent advances, it is clear that in the coming years, there will be further improvements in genetic testing for children with hearing loss. This is especially important given the recent progress in potential genetic therapy for hearing loss, as any genetic therapy first requires a confirmed genetic diagnosis.
References
Mehra S, Eavey RD, Neck DKJO-H. The epidemiology of hearing impairment in the United States: newborns, children, and adolescents. Elsevier. 2009;140(4):461–72.
Morton CC, Nance WE. Newborn hearing screening—a silent revolution. N Engl J Med. 2006;354(20):2151–64.
Qi S, Mitchell RE. Large-scale academic achievement testing of deaf and hard-of-hearing students: past, present, and future. J Deaf Stud Deaf Educ. 2011;17(1):1–18.
Teasdale TW, Sorensen MH. Hearing loss in relation to educational attainment and cognitive abilities: a population study: Hipoacusia en relación con los logros educativos y las habilidades cognitivas: Estudio en una población. Int J Audiol. 2007;46(4):172–5.
Fitzpatrick EM, Gaboury I, Durieux-Smith A, Coyle D, Whittingham J, Nassrallah F. Auditory and language outcomes in children with unilateral hearing loss. Hear Res. 2019;372:42–51.
Lieu JEC. Speech-language and educational consequences of unilateral hearing loss in children. Arch Otolaryngol - Head Neck Surg. 2004;130(5):524–30.
Lieu JE, Tye-Murray N, Karzon RK, Piccirillo JF. Unilateral hearing loss is associated with worse speech-language scores in children. Pediatrics. 2010;125(6):e1348–55.
Moore DR, Zobay O, Ferguson MA. Minimal and mild hearing loss in children: association with auditory perception, cognition, and communication problems. Ear Hear. 2019;1.
Roland L, Fischer C, Tran K, Rachakonda T, Kallogjeri D, Lieu JE. Quality of life in children with hearing impairment: systematic review and meta-analysis. Otolaryngol Neck Surg. 2016;155(2):208–19.
Loy B, Warner-Czyz AD, Tong L, Tobey EA, Roland PS. The children speak: an examination of the quality of life of pediatric cochlear implant users. Otolaryngol--Head Neck Surg Off J Am Acad Otolaryngol-Head Neck Surg. 2010;142(2):247–53.
Yoshinaga-Itano C, Sedey AL, Wiggin M, Mason CA. Language outcomes improved through early hearing detection and earlier cochlear implantation. Otol Neurotol. 2018;39(10):1256–63.
Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. Jama. 1999;281(23):2211–6.
Hilgert N, Smith RJ, Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat Res Mutat Res. 2009;681(2–3):189–96.
Shearer AE, Shen J, Amr S, Morton CC, Smith RJ. A proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet Med Off J Am Coll Med Genet. 2019;1–17.
Shearer AE, DeLuca AP, Hildebrand MS, Taylor KR, Gurrola J, Scherer S, et al. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc Natl Acad Sci. 2010;107(49):21104–9.
Shearer AE, Smith RJH. Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol – Head Neck Surg. 2015;153(2):175–82.
Shearer AE, Hildebrand MS, Sloan CM, Smith RJH. Deafness in the genomics era. Hear Res. 2011;282(1–2):1–9.
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135(4):441–50.
Nishio S, Usami S. Deafness gene variations in a 1120 nonsyndromic hearing loss cohort: molecular epidemiology and deafness mutation spectrum of patients in Japan. Ann Otol Rhinol Laryngol. 2015;124(1_suppl):49S–60S.
Wentland CJ, Ronner EA, Basonbul RA, Pinnapureddy S, Mankarious L, Keamy D, et al. Utilization of diagnostic testing for pediatric sensorineural hearing loss. Int J Pediatr Otorhinolaryngol. 2018;111:26–31.
Tekin D, Tutar E, Akay HO, Blanton S, Foster J, Tekin M. Comprehensive genetic testing can save lives in hereditary hearing loss. Clin Genet. 2015;87(2):190–1.
Sheppard S, Biswas S, Li MH, Jayaraman V, Slack I, Romasko EJ, et al. Utility and limitations of exome sequencing as a genetic diagnostic tool for children with hearing loss. Genet Med. 2018;20(12):1663–76.
Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–55.
Downie L, Halliday J, Burt R, Lunke S, Lynch E, Martyn M, et al. Exome sequencing in infants with congenital hearing impairment: a population-based cohort study. Eur J Hum Genet. 2019;1–10.
Liu P, Meng L, Normand EA, Xia F, Song X, Ghazi A, et al. Reanalysis of clinical exome sequencing data. N Engl J Med. 2019;380(25):2478–80.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(1):11–0.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology. 2016;123(5):1143–50.
Hao Z, Fu D, Ming Y, Yang J, Huang Q, Lin W, et al. Large scale newborn deafness genetic screening of 142,417 neonates in Wuhan, China. Bandapalli OR, editor. PLoS One. 2018;13(4):e0195740–15.
He X, Li X, Guo Y, Zhao Y, Dong H, Dong J, et al. Newborn screening of genetic mutations in common deafness genes with bloodspot-based gene chip array. Am J Audiol. 2018;27(1):57–11.
Wang Q-J, Zhao Y-L, Rao S-Q, Guo Y-F, He Y, Lan L, et al. Newborn hearing concurrent gene screening can improve care for hearing loss: a study on 14,913 Chinese newborns. Int J Pediatr Otorhinolaryngol. 2011;75(4):535–42.
Shearer AE, Eppsteiner RW, Booth KT, Ephraim SS, Gurrola J, Simpson A, et al. Utilizing ethnic-specific differences in minor allele frequency to recategorize reported pathogenic deafness variants. Am J Hum Genet. 2014;95(4):445–53.
Liming BJ, Carter J, Cheng A, Choo D, Curotta J, Carvalho D, et al. International pediatric otolaryngology group (IPOG) consensus recommendations: hearing loss in the pediatric patient. Int J Pediatr Otorhinolaryngol. 2016;90:251–8.
Alford RL, Arnos KS, Fox M, Lin JW, Palmer CG, Pandya A, et al. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet Med. 2014;16(4):347–55.
Jayawardena ADL, Shearer AE, Smith RJH. Sensorineural Hearing Loss: A Changing Paradigm for Its Evaluation. Otolaryngol – Head Neck Surg. 2015.
Ross SA, Ahmed A, Palmer AL, Michaels MG, Sánchez PJ, Stewart A, et al. Newborn dried blood spot polymerase chain reaction to identify infants with congenital cytomegalovirus-associated sensorineural hearing loss. J Pediatr. 2017;184:57–61.
Gantt S, Dionne F, Kozak FK, Goshen O, Goldfarb DM, Park AH, et al. Cost-effectiveness of universal and targeted newborn screening for congenital Cytomegalovirus infection. JAMA Pediatr. 2016 Dec 1;170(12):1173–80.
Fowler KB, McCollister FP, Sabo DL, Shoup AG, Owen KE, Woodruff JL, et al. A targeted approach for congenital cytomegalovirus screening within newborn hearing screening. Pediatrics. 2017;139(2):e20162128.
Lanzieri TM, Chung W, Flores M, Blum P, Caviness AC, Bialek SR, et al. Hearing loss in children with asymptomatic congenital cytomegalovirus infection. Pediatrics. 2017;139(3):e20162610.
Meyer L, Sharon B, Huang TC, Meyer AC, Gravel KE, Schimmenti LA, et al. Analysis of archived newborn dried blood spots (DBS) identifies congenital cytomegalovirus as a major cause of unexplained pediatric sensorineural hearing loss. Am J Otolaryngol. 2017;38(5):565–70.
Peterson J, Nishimura C, Smith RJH. Genetic testing for congenital bilateral hearing loss in the context of targeted cytomegalovirus screening. Laryngoscope. 2020;lary.28536.
Chen JX, Kachniarz B, Shin JJ. Diagnostic yield of computed tomography scan for pediatric hearing loss: a systematic review. Otolaryngol Neck Surg. 2014;151(5):718–39.
Kachniarz B, Chen JX, Gilani S, Shin JJ. Diagnostic yield of MRI for pediatric hearing loss: a systematic review. Otolaryngol Neck Surg. 2015;152(1):5–22.
Licameli G, Kenna MA. Is computed tomography (CT) or magnetic resonance imaging (MRI) more useful in the evaluation of pediatric sensorineural hearing loss? Laryngoscope. 2010;120(12):2358–9.
Joint Committee on Infant Hearing. Year 2007 position statement: principles and guidelines for early hearing detection and intervention programs. Pediatrics. 2007;120(4):898–921.
Funding
Dr. Kenna receives support from NIDCD DC015052.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical collection on Hearing Loss in Children
Rights and permissions
About this article

Cite this article
Shearer, A.E., Kenna, M. A Practical Approach to Genetic Testing for Pediatric Hearing Loss. Curr Otorhinolaryngol Rep 8, 250–258 (2020). https://doi.org/10.1007/s40136-020-00296-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40136-020-00296-5