
Abstract
Purpose of Review
The purpose of this review was to compare existing strategies for evaluation of complex paediatric patients with newer techniques. Comparative genomic hybridization (CGH) array is the currently accepted first tier genetic test in the evaluation of a pediatric patient with complex physical and developmental anomalies. CGH provides an answer in only 15–20 % cases, and further genetic testing is required in the majority of cases. This has previously involved sequential single-gene tests, with low yield, significant costs and delay in diagnosis.
Summary of Recent Findings
New genetic techniques allowing massively parallel sequencing of multiple genes are becoming a part of medical practice as they provide a reduction in cost and time. Current medical practice supports the use of limited genomic testing—‘gene panels’ and ‘clinical exomes’, as a second tier approach after CGH array testing. These approaches have already been shown to improve the diagnostic yield providing an answer for an additional 25 % of patients. Ultimately, it is likely that whole genome sequencing as a single genomic test could replace CGH array and more restricted genomic tests, as research experience is translated into medical practice. Several factors need to be overcome to make this a reality to ensure equitable access to a reliable test with appropriate diagnostic interpretation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Children with unexplained developmental disability and other congenital anomalies are a common referral to pediatric clinics. Single-gene testing has had a low yield despite good clinical characterization of individual patients [1]. The advent of human genome sequencing has disproved the concept of single gene causing a single genetic disorder. A variation in one of the several genes may cause the same clinical phenotype—for example, there are at least seven genes associated with Noonan syndrome [2]. Conversely, an identical genetic change may be associated with several different neurodevelopmental and behavioural disorders—autism, developmental disability, mental retardation and schizophrenia and other adult-onset mental health problems may all be associated with a change in the same gene [3–5]. These same variants may also be present in parents who have no unusual clinical features.
Comparative genomic hybridization (CGH) (Table 1—Glossary) was the first detailed ‘whole genome’ study to be introduced, allowing detection of alterations in chromosome content across the whole genome without prior knowledge of the chromosome involved. Newer genetic techniques, collectively called next generation sequencing (NGS) to differentiate from the original Sanger method of single base-pair sequencing, are transitioning from research lab to standard medical care. NGS techniques, or more appropriately massively parallel sequencing (MPS), allow for the screening of multiple genes (up to a whole genome) in a single test. These technologies can be considered in three main groups of increasing complexity and capacity:
-
(1)
Targeted multiple gene panels.
-
(2)
Whole exome sequencing (WES).
-
(3)
Whole genome sequencing (WGS).
This review will examine the potential for diagnostic incorporation of these technologies in the context of the evaluation of a paediatric patient with complex developmental and other anomalies, compared to existing techniques including CGH and single-gene testing (usually done by Sanger sequencing).
These new technologies are sometimes described as ‘hypothesis free’ or ‘data driven’ because no knowledge of the disease-causing gene is required before testing. However, it is imperative that detailed clinical assessment and family history have been undertaken by the requesting clinician in order to assist in the interpretation of complex and sometimes uncertain results.
CGH Array
Standard G-banded chromosome test (karyotyping) has a resolution of 5–10 million bases (5–10 Mb) and detects chromosomal alterations in ~5 % of individuals with unexplained intellectual disability [6]. Molecular karyotyping methods, such as microarray-based CGH (array CGH; aCGH), can detect submicroscopic chromosome alterations, such as duplications and deletions within a chromosome, at a resolution of ~100 kilobases (kb). CGH is able to detect mosaicism for whole chromosome trisomy, but is not able to detect chromosome rearrangements, such as inversions and balanced chromosome translocations, that do not alter copy number. Karyotype should be requested in this situation. CGH may also miss variants located in parts of the genome which are not well covered by the array.
CGH detectable chromosome imbalances, called ‘copy number variants’ (CNVs), are common in the general population, but specific rare CNVs are an important cause of developmental disability and congenital anomalies. Various studies have shown an increased diagnostic yield from CGH for children with developmental disability [1, 7–9] who had a normal karyotype test. This yield is highest when associated with other congenital anomalies or physical differences such as “dysmorphic” facial features or disturbances in pre- and postnatal growth [2, 9]. 15–20 % children with intellectual disability will have a pathogenic CNV detected on CGH [3, 4, 8]. CGH has been shown to be a robust technique with clinical and analytical validity and is now recommended as the first-line test replacing the traditional karyotype, except in special cases [6, 10–12].
As with all genomic tests, there are limitations including a false positive rate of approximately 5 % due to identification of a copy number variant of uncertain significance (VUS) which requires the need for parental testing, adding to costs [7] and increasing uncertainty. Guidelines have been developed for interpretation and reporting of CNV’s by the American College of Medical Geneticists [12, 13] and equivalent bodies in Europe [14] and Australasia [15]. Diagnostic reports usually categorise an identified CNV into one of the following categories:
-
Pathogenic (clearly known to be associated with a clinical phenotype).
-
Likely pathogenic—could explain the phenotype but more studies are required.
-
Benign—known to exist in the general population without obvious effect.
-
Variant of uncertain significance—insufficient information is available to classify the CNV.
Parental studies are required to improve the accuracy of reporting in many cases, adding costs and time. Recently, whole exome sequencing (WES) has been shown to provide further clarification in some cases, by identifying additional pathogenic variants not detected by CGH [16]. CNVs may be de novo or familial and some are associated with a range of consequences in cognitive and behavioural development within a family whose members each carry the same CNV [4]. These recurrent CNVs are sometimes called ‘susceptibility loci’ as their clinical effect is modulated by other inherited factors and is therefore modulated by family history [3, 4].
For the remaining 80 % of children with no pathogenic variant detected by CGH, further sequential single-gene testing has often been required, usually directed by clinical examination of the child. This has proven to have a low diagnostic yield, and to be costly and emotionally difficult for families because of the extended time to diagnosis [1, 17]. Sanger sequencing has been the gold standard for gene sequencing but is costly and time consuming due to the nature of the reactions—sequencing a single gene in a single patient, single base pairs at a time. This has limited the accessibility of genetic testing for many families.
Massively Parallel Sequencing (MPS)
MPS has changed the diagnostic landscape for genetic disease, by allowing the ability to rapidly sequence multiple genes in parallel. The technique involves fragmenting a patient’s DNA and then sequencing each fragment multiple times in parallel. The data are then collated and curated into a consensus sequence for the patient, and variants are annotated by comparing to a reference sequence (for more details, see Muzzey et al. and Biesecker et al. [18∙, 19∙]). The literature generally discusses the quality of sequencing data in terms of depth of coverage, or the number of times a particular segment of the genome is sequenced.
Exome-Based Sequencing
All exome-based sequencing techniques rely on the capture and sequencing of only the exons, or protein coding region, of the genome [20]. This reduces the volume of raw sequencing data significantly (10–12 Gb of sequencing data per exome compared to ~140 Gb per whole genome) since protein coding regions represent <2 % of the whole genome [21, 22]. To date, approximately 85 % of known disease-causing genetic changes have been identified in the exons [23]. This capture step can potentially introduce bias, particularly in homologous or repetitive segments of the genome [21] and results in less-uniform depth of coverage. Exome-based techniques also have difficulty in detecting large deletions or structural variants such as rearrangements [21].
Clinical Exomes and Targeted Gene Panels
Clinical Exomes focus on the capture and sequencing of genes that are known to be disease causing or clinically relevant, rather than all known coding regions. The benefit of this more targeted approach is that this mode of testing is often more cost efficient for laboratories and is more tailored to a diagnostic rather than research setting, where there is limited resources to pursue variants in novel or little-known genes.
Gene panels also offer a targeted diagnostic approach in which only a set of genes associated with a particular phenotype are either sequenced or analysed. This approach could be considered in a child with specific phenotypic features that may indicate a particular group of diseases or genes, such as, for example, when investigating overgrowth features or hypertrophic cardiomyopathy. This mode of testing can consume more time and be cost efficient in these groups of patients. Diagnostic rates in cohorts of patients with a specific phenotype are variable depending on the phenotype and breadth of genes sequenced. In a cohort of 400 patients with severe developmental delay and/or early onset seizures, a diagnosis was made in 18 % utilising a panel of 46 known genes sequenced via an exome-based platform in conjunction with chromosomal microarray [24]. The group noted that patients with more severe phenotype (seizure onset before 2 months age) were more likely to have a molecular diagnosis made [24].
Gene panels and Clinical Exomes are a cost-efficient mode of diagnostic sequencing, but given the pace in which gene discovery is occurring, there is the potential for these methods to become out-of-date. In addition, if the test method sequences targeted genes only, it is not possible to re-analyse data by extending the genes analysed, if a diagnosis is not made on the first round of testing.
Whole Exome Sequencing
WES sequences all coding regions of the genome. Diagnostic laboratories can then undertake analysis of variants over the entire coding region, or target initial analysis to particular gene sets, with the potential to widen the genes analysed [25].
The Deciphering Developmental Disorders (DDD) Study was a UK-based study of over 1000 children with undiagnosed developmental disorders (including intellectual disability or developmental delay, seizures, congenital heart defects and autism spectrum disorder) despite previous investigation [26∙]. The children underwent ‘trio’ WES (WES of affected child and both parents) and high-resolution microarray, in which all coding regions were sequenced and variants were filtered using parental results and according to phenotype. A diagnosis was made in 27 % of children, with the majority having de novo variants [26∙]. This study highlights the diagnostic power of WES, particularly when parental samples and detailed phenotypic information are available. Other groups have also demonstrated similar diagnostic rates of approximately 30 % in cohorts with undiagnosed complex disorders [25, 27–30].
A concern for health services is the cost of exome sequencing. Valencia et al. assessed the utility of WES in a paediatric cohort with a variety of primary diseases, including immunodeficiency, mitochondrial or neurological disorders and complex congenital anomalies [29]. Prior to inclusion in the study, most patients had undergone single-gene sequencing and chromosomal microarray, and approximately one-third had been investigated with a multi-gene panel without diagnosis. A diagnostic rate of 30 % was achieved with WES without trio analysis [29]. The authors discussed that in appropriate cases, early WES has utility in avoiding multiple costly investigations and in shortening the diagnostic odyssey for children and their families.
Detailed clinical phenotyping assists in appropriate test selection and improving diagnostic yield. A prospective study of 80 infants with clinical features suggestive of monogenic disease (multiple congenital abnormalities and dysmorphic features, skeletal dysplasia, neuro-metabolic conditions) demonstrated a 57.5 % diagnostic rate through singleton WES, compared with 13.75 % in infants who underwent standard investigation [31]. All 80 infants in the study underwent WES, and 21 of these 80 also underwent standard clinical testing in parallel [31]. In this study, clinicians compiled a list of candidate genes based on the patient’s phenotype, and variants were initially prioritised based on this list [31]. This study highlights the value of phenotyping and careful patient selection to increase diagnostic yield with sequencing. The authors discuss that the higher diagnostic yield in this study compared to others is likely due to early use of WES and the characteristics of the patients [31]. Other groups have also demonstrated that diagnostic yield appears to be higher in more ‘complex’ or higher acuity patients [32, 33].
As with all broad testing approaches, WES increases the potential of identifying incidental or secondary findings—findings that may have medical relevance for a patient but were not the reason for the testing. Laboratories vary in their policy regarding reporting of incidental findings but increasing consensus is developing. [34–36]. Some may elect to avoid analysis of particular genes, such as those that predict adult-onset disease, as suggested in some guidelines [35]. In addition to incidental findings, broad testing increases the possibility of identifying ‘variants of uncertain significance’ (VUS) that requires further characterisation and is not able to be used for predictive testing or family planning. In one study, VUS were identified in 86 % of patients, with 53 % needing follow-up with additional laboratory testing and genetic testing of other family members [25]. Consensus guidelines are also developing for the classification of genome variants identified during WES and WGS testing in a similar manner to that established for larger CNVs [37, 38]. These issues highlight the importance of adequate consent prior to any sequencing, in order to ensure that patients and families are aware of the possible uncertainty that genetic testing can bring.
Whole Genome Sequencing
WGS involves the sequencing of all coding and non-coding segments of the genome. This sequencing method therefore avoids the capture process of exome-based sequencing methods and is able to achieve more uniform coverage across the genome [39, 40]. WGS is also better able to detect structural variants (deletions, duplications and rearrangements) and sequence homologous regions of the genome than exome-based methods [41∙, 42].
WGS has been trialled in a group of 50 patients with severe intellectual disability (IQ < 50) who had previously been extensively investigated with targeted gene analysis, WES and chromosomal microarray without a molecular diagnosis being made [41∙]. Via trio-WGS, a diagnosis was made in 42 % of patients, with the majority having de novo variants [41∙]. The majority of the variants were detected in the coding region of the genome, and the group discussed that further investigation is required to understand the impact of non-coding variants on disease [41∙].
In addition to intellectual disability, WGS has also had impact in cohorts of patients with single organ disorders, for example eye disease, with a group demonstrating that WGS was able to make a diagnosis in some patients who had undergone WES without diagnosis, and detect additional disease-causing variants in those who had already had a molecular diagnosis made with WES [43]. Another group utilised singleton WGS in a cohort of patients with a recessive retinitis pigmentosa phenotype and was able to make a molecular diagnosis in half of patients [20]. The group also discovered a novel disease gene, which highlights the research power of WGS.
In addition to detecting single nucleotide variants (SNV), multiple groups have demonstrated that WGS is able to detect structural variants with breakpoints in non-coding regions, which would not have been detected by exome-based methods [20, 41, 43].
Some of the practical concerns regarding WGS are the processing and storage of the large amounts of data generated with sequencing of the whole genome (approximately 130 Gb of raw data) and the cost of the technique. Soden et al. performed a study in which 119 children with neurodevelopmental delay underwent trio WES or WGS [33]. Patients were selected for WES or WGS based on the acuity of their illness, with 15 families with infants in neonatal or paediatric intensive care undergoing rapid 50-h WGS [33]. A diagnosis was made in 45 % of families enrolled in the study. Of those who underwent rapid WGS, 75 % had a diagnosis made [33]. The high diagnostic rate in the subset of critically ill infants is likely a reflection of the acuity and early onset of their disease, and that they had undergone limited prior investigation. In the less acute group, the investigators estimated that families had already undergone $19,100 of testing prior to sequencing. They therefore calculated that genomic sequencing would be cost-effective for diagnosis at a cost of no more than $7640 per family [33]. The group also reported that WGS was able to make a diagnosis in a family that failed WES, after a variant was found in the coding region of a GC-rich gene that had poor coverage on WES [33]. This demonstrates the power of WGS in difficult-to-sequence regions of the genome. Interestingly, on review of the clinical data, the group reported that diagnosis was more likely to be made in children with a history of failure to thrive or intrauterine growth retardation [33].
The diagnostic potential in this and other studies is assisted by the ability to sequence trios, which can be costly in a diagnostic setting. Soden et al. also used clinico-pathologic software to translate patients' clinical features into lists of disease genes to initially analyse, and again highlights the value of detailed phenotypic information to aid diagnosis. Given the rapid advancement in the field over recent years, it is likely that the cost of all sequencing, including WGS, is likely to decline and therefore become more accessible.
As with WES, the potential for incidental findings and variants of uncertain significance is present with WGS and therefore requires careful genetic counselling and consenting prior to testing for all family members. In a recent Canadian study of 100 children referred for CGH, WGS was offered in parallel [39]. On average, two additional gene tests had been requested on each child at the time of referral for CGH. This included a targeted gene panel in 22 children. Results indicated WGS alone identified the cause of the disability in 34 % compared to 8–13 % for CGH. Of those who had simultaneous genetic testing, the identified gene sequence was absent from the panel tested in 17/22 cases, highlighting the limitation of targeted panels, which may not include all the genes associated with that specific clinical condition. Interestingly, WGS was also shown to be able to identify all the CNVs detected by CGH suggesting the possibility of WGS being a first tier test in the future. This study also highlighted the difficulties to be overcome when offering WGS to families. 2 % of patients had an additional medically actionable variant identified. Two diagnoses were missed—UPD14 (uniparental heterodisomy of chromosome 14) and a methylation abnormality in a Russell Silver Syndrome patient. In recruiting for the study, consecutive families were approached with 95/201 families declining to be involved in the WGS study including 35 % due to concerns about insurance implications of potential secondary findings; a further 35 % felt too overwhelmed with current medical complexity of their child and did not want any further information [39]. Obtaining informed consent is essential before proceeding with genomic testing.
Practical Considerations
An important consideration with any diagnostic genetic testing is the selection of testing laboratory. Guidelines recommend the use of an accredited diagnostic laboratory for diagnostic testing [44]. Given the challenges in interpretation of variants, consideration should also be made regarding the expertise of the laboratory to interpret variants related to the patient’s phenotype. For example, a particular laboratory may have expertise in interpreting variants in patients with cardiomyopathy and thus may be better suited for testing of this group of patients. Several online databases may assist in these decisions (Table 2).
The decision-making process that laboratories undertake to interpret variants is complex and requires expertise. Depending on the test ordered, a patient may have numerous potential variants identified that require individual analysis by the laboratory team. This includes accessing large population datasets, such as the ExAC Database [45], to assess the frequency of the variant in groups of relatively well patients. Numerous disease databases also exist which compile variants from patients known to have a particular disease [46]. These databases can also be consulted to assess variant pathogenicity. Laboratories also use in silico analysis tools—utilising computational models that can predict the pathogenicity of variants with varying levels of certainty [47]. Guidelines suggest that multiple criteria must be satisfied to assess a variant as pathogenic or likely pathogenic [48]. If insufficient evidence is present, these will be classified as VUS. Complexity increases when two potentially pathogenic variants are found in two different genes, can be reported in hypertrophic cardiomyopathy [49]. In these instances, the clinician relies heavily on laboratories with expertise in analysis of particular disease groups to interpret these results.
Finally, as with all aspects of medicine, best practice is dependent on multidisciplinary collaboration. Given the complexity of test selection and interpretation, potential for incidental findings and requirement for genetic counselling, exome and genome sequencing should be considered in conjunction with a clinical genetics service. Equally, given the complexity and vital importance of clinical phenotyping, clinical and laboratory geneticists need to have strong links with treating physicians.
Conclusion
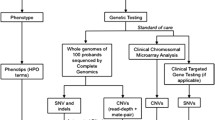
Recent advances in genetic sequencing techniques have dramatically altered the diagnostic pathway for children with complex disease. The ability to broadly analyse multiple genes has also expanded disease phenotypes, and allowed diagnosis of diseases for which patients have atypical clinical features [33, 39]. This shows that the evolving sequencing technologies have not only increased diagnostic rates but also our understanding of existing disease. Increase in testing options does increase the challenge for the clinician in appropriate test selection for each patient. The literature demonstrates that the yield with broad testing, such as WES or WGS, is increased if this testing is performed earlier in the diagnostic pathway. Importantly, earlier diagnosis reduces periods of uncertainty for families and avoids a prolonged diagnostic odyssey. Practically, this has to be weighed against the cost of such broad testing and the ability for health services to cover these costs [50]. Given the considerations, CGH array remains a cost-effective first-line test (Fig. 1). As technology improves, these costs for WES and WGS should reduce.

Algorithm for assessment of a complex child. Algorithm to guide genetic testing in the assessment of a complex child. WES whole exome sequencing, WGS whole genome sequencing, dels deletions, dups duplications
More importantly, consideration must be given to implications of testing both to the child and the extended family. Genetic testing for adult-onset disorders is not recommended in children and future implications of testing should be considered carefully, particularly given the potential to identify incidental findings or variants of uncertain significance [19, 34, 35].
For this reason, genetic counselling and adequate consent are essential before considering any genetic testing (Fig. 1).
Despite the complexity of genetic testing that is available to the clinician, the fundamental principles for diagnostic testing remain—that testing should be considered if the result is likely to alter management for the patient or family. Included in this should be consideration of the benefit of resolution of uncertainty when a clear diagnosis is made, even in conditions for which there are limited treatment options. Finally, regardless of the testing approach chosen, diagnostic yield is improved with detailed phenotyping that allows selection of an appropriate test and tailored analysis of variants in the context of the specific patient.
References
Papers of particular interest, published recently, have been highlighted as: ∙ Of Importance
Lynch SA. What price a diagnosis? Dev Med Child Neurol. 2011;53:971.
Aoki Y, Niihori T, Inoue S-I, Matsubara Y. Recent advances in RASopathies. J Hum Genet. 2015;61:33–9.
Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL, et al. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet. 2011;155:2386–96.
Finucane B, Challman TD, Martin CL, Ledbetter DH. Shift happens: family background influences clinical variability in genetic neurodevelopmental disorders. Genet Med. 2015;18:302–4.
Vissers L, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17:9–18.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64.
Bejjani BA, Shaffer LG. Application of array-based comparative genomic hybridization to clinical diagnostics. J Mol Diagn. 2006;8:528–33.
Vissers LELM, de Vries BBA, Veltman JA. Genomic microarrays in mental retardation: from copy number variation to gene, from research to diagnosis. J Med Genet. 2010;47:289–97.
de Vries BBA, Pfundt R, Leisink M, Koolen DA, Vissers LELM, Janssen IM, et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–16.
Xiang B, Li A, Valentin D, Nowak NJ, Zhao H, Li P. Analytical and clinical validity of whole-genome oligonucleotide array comparative genomic hybridization for pediatric patients with mental retardation and developmental delay. Am J Med Genet. 2008;146A:1942–54.
Manning M, Hudgins L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12:742–5.
Kearney HM, South ST, Wolff DJ, Lamb A, Hamosh A, Rao KW. American College of Medical Genetics recommendations for the design and performance expectations for clinical genomic copy number microarrays intended for use in the postnatal setting for detection of constitutional abnormalities. Genet Med. 2011;13:676–9.
South ST, Lee C, Lamb AN, Higgins AW, Kearney HM. ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: revision 2013. Genet Med. 2013;15:901–9.
Hanemaaijer NM, Sikkema-Raddatz B, van der Vries G, Dijkhuizen T, Hordijk R, van Essen AJ, et al. Practical guidelines for interpreting copy number gains detected by high-resolution array in routine diagnostics. Eur J Hum Genet. 2011;20:161–5.
Bruno D, Beddow R, Caramins M, Fagan K, Greville W, Harraway J, et al. Interpreting clinical microarray genomic data in 2012: what have we learnt and what challenges remain? Curr Top Genet. 2012;5:67–79.
Giorgio E, Ciolfi A, Biamino E, Caputo V, Di Gregorio E, Belligni EF, et al. Whole exome sequencing is necessary to clarify ID/DD cases with de novo copy number variants of uncertain significance: two proof-of-concept examples. Am J Med Genet. 2016;. doi:10.1002/ajmg.a.37649.
Gogarty B. Parents as Partners: a report and Guidelines on the investigations of children with developmental delay; by parents for professionals. Public health Genetics Unit, Cambridge Genetics Knowledge Park, 2006. Available at SSRN: http://ssrn.com/abstract=1796762.
∙ Muzzey D, Evans EA, Lieber C. Understanding the Basics of NGS: From Mechanism to Variant Calling. Curr Genet Med Rep. 2015;3:158–65. Review of technical aspects of Massively Parallel Sequencing techniques.
∙ Biesecker LG, Green RC. Diagnostic Clinical Genome and Exome Sequencing. N Engl J Med. 2014;370:2418–25. Review of technical aspects of Massively Parallel Sequencing techniques.
Nishiguchi K, Tearle R, Liu Y, Oh E, Katsanis N, Rivolta C. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structuralchanges and NEK2 as a new disease gene. Proc Natl Acad Sci USA. 2013;10:16139–44.
Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge M, Jhangiani S, Buhay C, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Gen Med. 2013;5:57.
Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–6.
Mahdieh N, Rabbani B. An overview of mutation detection methods in genetic disorders. Iran J Pediatr. 2013;23:375–88.
Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet. 2016;53:310–7.
Shashi V, McConkie-Rosell A, Schoch K, Kasturi V, Rehder C, Jiang YH, et al. Practical considerations in the clinical application of whole-exome sequencing. Clin Genet. 2015;89:173–81.
∙ Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. The Lancet. 2015;385:1305–14. Major study of exome sequencing in Development Disorders.
de Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–11.
Valencia CA, Husami A, Holle J, Johnson JA, Qian Y, Mathur A, et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front Pediatr. 2015;3:121.
Monroe GR, Frederix GW, Savelberg SMC, de Vries TI, Duran KJ, van der Smagt JJ, et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genetics in Medicine. 2016:1–8.
Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;. doi:10.1038/gim.2016.1.
Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 2015;314:895.
Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168.
van El CG, Cornel MC, Borry P, Hastings RJ, Fellmann F, Hodgson SV, et al. Whole-genome sequencing in health care. Eur J Hum Genet. 2013;21:S1–5.
Watson M. Incidental findings in clinical genomics: a clarification. Genetics in Medicine. 2013;15:664–6.
Hehir-Kwa JY, Claustres M, Hastings RJ, van Ravenswaaij-Arts C, Christenhusz G, Genuardi M, et al. Towards a European consensus for reporting incidental findings during clinical NGS testing. Eur J Hum Genet. 2015;23:1601–6.
Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am J Hum Genet. 2016;98:1067–76.
Jarvik GP, Browning BL. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Hum Genet. 2016;98:1077–81.
Stavropoulos DJ, Merico D, Jobling R, Bowdin S, Monfared N, Thiruvahindrapuram B, et al. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. Gen Med. 2016;1:15012.
Meienberg J, Bruggmann R, Oexle K, Matyas G. Clinical sequencing: is WGS the better WES? Hum Genet. 2016;135:359–62.
∙ Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BWM, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7. Major study of Whole Genome Sequencing in intellectual disability.
Mallawaarachchi AC, Hort Y, Cowley MJ, McCabe MJ, Minoche A, Dinger ME, et al. Whole-genome sequencing overcomes pseudogene homology to diagnose autosomal dominant polycystic kidney disease. Eur J Hum Genet. 2016;. doi:10.1038/ejhg.2016.48.
Ellingford J, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, et al. Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology. 2016;123:1143–50.
Matthijs G, Souche E, Alders MEL, Corveleyn A, Eck S, Feenstra I, et al. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet. 2016;24:2–5.
ExAC Browser [Internet]. Broad Institute; [cited 2016 Jun 19]. http://exac.broadinstitute.org.
The Human Gene Mutation Database [Internet]. Institute of Medical Genetics in Cardiff; [cited 2016 Jun 19]. http://www.hgmd.cf.ac.uk/ac/index.php.
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–37.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years. JAC. 2012;60:705–15.
Beale S, Sanderson D, Sanniti A, Dundar Y, Boland A. A scoping study to explore the cost-effectiveness of next-generation sequencing compared with traditional genetic testing for the diagnosis of learning disabilities in children. Health Technol Assess. 2015;19:1–90.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
Amali Mallawaarachchi and Felicity Collins declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Genetics.
Rights and permissions
About this article

Cite this article
Mallawaarachchi, A., Collins, F. Testing the Complex Child: CGH Array, WES, Clinical Exome, WGS. Curr Pediatr Rep 4, 155–163 (2016). https://doi.org/10.1007/s40124-016-0111-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-016-0111-6