
Abstract
Severe combined immunodeficiency (SCID) is a life-threatening disease caused by a heterogeneous group of genetic defects. It is characterized by profound defects of T-cell development, also affecting B and NK cells in some cases. Since the first molecular identification of a causal gene for SCID in 1985, 14 more molecular causes have been identified in patients with a classical SCID phenotype, with no T cells. Some genetic defects specifically block lymphocyte ontogeny, whereas others affect T-cell function and a few cause extra-hematopoietic alterations with a rare complex phenotype. Over the last 15 years, several new causal genes have been identified in patients with low T-cell counts and impaired T-cell function. Patients with a clinical SCID phenotype with normal numbers of dysfunctional T cells have also recently been reported. This last condition is described immunologically as combined immunodeficiency. These discoveries have expanded the complexity and difficulties of molecular characterization in patients with a clinical SCID phenotype. Studies of these disorders have increased our understanding of the role of single-gene products in the development, differentiation, and function of the immune system in humans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human primary immunodeficiencies (PIDs) comprise a broad group of inherited disorders characterized by developmental or functional defects of myeloid or lymphoid hematopoietic cells, and of non-hemapoietic cells involved in protective immunity [1, 2••]. Severe combined immunodeficiency diseases (SCID) are a heterogeneous group of PIDs caused by defects in T-lymphocyte development, with or without alterations to the development of B lymphocytes, NK lymphocytes, or myeloid cells [3]. The first published report of SCID can be traced back to 1950, when this condition was described in two siblings with profound lymphopenia [4]. A second report described thymic dysplasia and hypogammaglobulinemia in other SCID patients in 1958 [5]. Patients with SCID present recurrent infections caused by opportunistic microbes (such as Pneumocystis jiroveci) viruses, bacteria or fungi, during the first year of life [6]. On physical examination, these patients generally lack lymphatic tissue and no thymus shadow is detectable on chest X-ray [3].
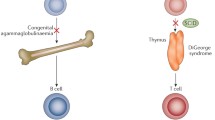
SCID patients generally present profound lymphopenia, with very low T-cell counts, typically below 500/µL, if indeed these cells can be detected at all. The immunophenotyping of T, B, and NK cells can improve the definition of SCID phenotype in patients, guiding subsequent genetic studies [3]. Four groups of SCIDs can be defined on the basis of the presence or absence of various lineages of lymphocytes: (i) SCID with no T, B, and NK cells (T−B−NK−), (ii) SCID with no T and NK cells (T−B+NK−) but with B cells, (iii) SCID with no T and B cells but with NK cells (T−B−NK+) and (iv) SCID with no T cells but with NK and B cells (T−B+NK+) (Table 1). Another variant of SCID is Omenn syndrome which may have early or delayed manifestations, and refers to a subset of infants in whom there is oligoclonal proliferation of dysregulated autologous T cells which lead to generalized erythroderma and desquamation of the skin, lymphadenopathy, splenomegaly, eosinophilia, and elevation of immunoglobulin E. Analyses of thymopoiesis based on T-cell receptor excision circles (TRECs) can be used for diagnostic purposes, particularly for the screening of neonates [7•] (see Kwan et al., review in this series). We review here SCID disorders and their molecular etiologies, mutations in the genes encoding cytokine receptors, key molecules in V(D)J recombination, pre-T-cell receptor (TCR) and TCR compounds, metabolic disorders, or impairment of the thymic development of T cells. Finally, we discuss a group of patients with a clinical SCID phenotype but no significant T-cell lymphopenia (table). These patients have normal numbers of T cells, but these cells are not functional, due to a T-cell activation defect (Fig. 1).

Genes affecting lymphoid development and function in SCID
T−B−NK−SCIDs
Adenosine Deaminase Deficiency
The first etiology of autosomal recessive (AR) T−B−NK−SCID was identified by Giblett et al. in 1972, who showed that these patients lacked the adenosine deaminase (ADA) enzyme [8]. The gene encoding ADA was mapped to chromosome 20q13.2–11 and cloned [9]. The ADA deficiency (MIM 102700) caused by mutations of this gene results in the marked accumulation of adenosine, 20-deoxyadenosine and 20-O-methyladenosine, and an absence of detectable ADA activity [10] This is accompanied by an intracellular accumulation of dATP. The first homozygous ADA mutation was reported in 1985 [11] and the first compound heterozygous mutations were identified in 1986, in one patient with ADA deficiency [12]. Since then, over 50 deleterious mutations have been identified in ADA-deficient patients [10]. Typically, ADA-deficient patients have profound lymphopenia, with mean absolute lymphocyte counts of less than 500/µL and a deficiency of all three types of lymphocytes (T−B−NK−SCID) [10]. ADA-deficient patients also display the skeletal abnormalities of chondro-osseous dysplasia, a flaring of the costochondral junctions and a bone-in-bone abnormality of the vertebral bodies [10] There are also other non-immunological defects, such as cognitive and behavioral abnormalities, deafness, and pulmonary defects. Milder forms of this condition have been reported, potentially delaying the diagnosis of immunodeficiency with CD4 T lymphopenia, even in adult patients [13]. ADA deficiency accounts for 17 % of SCID patients in North America and 4 % of SCID patients in France [3, 14] and the exact incidence is very dependent on the specific population. In patients with T−B−NK−SCID, ADA activity or dATP accumulation can be assessed before genetic testing, in whole-blood cells or isolated leukocytes, if the patient has received a transfusion of red blood cells.
Adenylate Kinase 2 Deficiency
Reticular dysgenesis is an AR SCID characterized by a premature arrest of differentiation in the myeloid lineage and impaired lymphoid maturation, associated with sensorineural deafness. It was first clinically described in 1959 [15]. Patients have a T−B low NK-phenotype and neutropenia [16, 17]. Linkage mapping by two independent groups in 2009 led to the demonstration that mutations of the AK2 gene were responsible for reticular dysgenesis [18, 19]. The AK2 gene encodes the mitochondrial energy metabolism enzyme adenylate kinase 2 (AK2). AK2 deficiency (OMIM 103020) causes the premature apoptosis of myeloid progenitor cells [18, 19]. Fifteen deleterious mutations have been identified in AK2-deficient patients. AK2-deficient patients display profound lymphopenia with deficiencies of all three types of lymphocytes, with no T cells (T−B±NK−SCID), neutropenia and, occasionally, anemia, and/or thrombocytopenia [10]. Bone marrow analysis shows a complete absence of mature myeloid cells [10]. This PID highlights the key role of the AK2 protein in hematopoiesis and is the first example of a human PID causally linked to energy metabolism and classified as a mitochondriopathy.
T−B+NK−SCIDs
IL-2 Receptor γc Deficiency
SCID X-linked (XL) was first described in 1958 [5]. Patients with SCID-XL lack circulating T and NK cells, but have normal or higher than normal numbers of B lymphocytes (T−B+NK−). In 1987, the causal gene for SCID-XL was mapped, by linkage analysis, to the proximal long arm of the X chromosome [20]. In 1993, two research groups determined that the IL2RG gene, which encodes the common gamma chain (γc) of the IL-2 receptor, was located on the proximal long arm of the X chromosome and identified the first deleterious hemizygous mutation in a boy with SCID-XL (OMIM 300400) [21, 22],. The disease-causing gene encodes the common cytokine receptor γ-chain (γc), which is essential for the response to at least six cytokines: interleukin (IL)-2, IL-4, IL-7, IL-9, IL-15, and IL-21 [22, 23]. Several hundred amorphic or hypomorphic IL2RG mutations have been reported [3, 10, 24, 25]. SCID-XL is the most common form of SCID, accounting for 25–46 % of SCID cases [3, 14, 26, 27]. In male patients with the T−B+NK−SCID phenotype, γc (CD132) expression on B cells should be assessed before genetic testing as the absence of γc expression confirms the diagnosis. After successful allogeneic hematopoietic stem cell transplantation (HSCT), γc-deficient patients have been found to have a higher than normal incidence of skin infections caused by human papilloma virus (HPV) during long-term follow-up, suggesting a role of γc-dependent cytokine receptors in the immunity of epithelial cells [28].
JAK3 Deficiency
AR T−B+NK−SCID, the immunological and clinical features of which are indistinguishable from those associated with γc deficiency, was first reported in 1993 [26]. JAK3 is a protein kinase of the Janus kinase family. As the only signaling molecule known to be required for signal transduction through γc-containing cytokine receptors, it was identified as a strong candidate gene for AR T−B+NK−SCID. Mutations of the JAK3 gene, encoding an intracellular tyrosine kinase, were first identified in 1995, in patients with AR SCID [29, 30]. More than 30 patients with JAK3 deficiency (OMIM 600173) have since been identified [10, 29–37]. JAK3 deficiency accounts for 10–18 % of all SCID cases [3, 14, 27]. A few patients with milder clinical phenotypes have been reported, with missense mutations or small in-frame deletions of the JAK3 gene [10, 35, 36]. These patients are able to develop autologous T cells due to residual JAK3 expression. Thus, JAK3 mutations are associated with AR T−B+NK−SCID, or, more rarely, with combined immunodeficiency (CID). Following successful allogeneic HSCT, JAK3-deficient patients have a higher frequency of skin HPV infections than the general population during long-term follow-up, as reported for γc-deficient SCID-XL patients, suggesting that NK cells or γc/JAK3-dependent signaling in keratinocytes may play a role in anti-HPV immunity [28].
T−B−NK+SCIDs
RAG1 and RAG2 Deficiencies
AR T−B−NK+SCID patients were first reported in 1988 [38]. A recombinase deficiency was then characterized in these T−B−NK+SCID patients, partly analogous to the defect observed in SCID mice [39]. RAG1 (OMIM 179615) and RAG2 (OMIM 179616) deficiencies were identified by applying a candidate gene strategy once the homologous genes had been cloned in mice, based on the analogous phenotypes of Rag1- and Rag2-knockout mice and some patients with AR T−B−NK+SCID [40]. Recombination-activating genes 1 and 2 (RAG1 and RAG2) encode enzymes required for the initiation of V(D)J recombination in T and B cells [41]. The first six cases of RAG deficiency were detected in single-strand conformation polymorphism assays and direct sequencing then identified homozygous or compound heterozygous mutations in the RAG1 and RAG2 genes, respectively [42]. RAG1 and RAG2 deficiencies account for 10 % of all SCID cases in North America and 22 % in Europe [3, 14]. In addition to causing the SCID phenotype, hypomorphic mutations of the RAG1 and RAG2 genes lead to partially impaired V(D)J recombination activity, resulting in Omenn’s syndrome or a leaky or atypical CID phenotype [10, 43] although Omenn’s syndrome can arise from hypomorphic mutations in other SCID causing genes. Flow cytometry analysis of the B-cell precursor compartment in bone marrow can be helpful, because typical SCID patients with RAG1 or RAG2 deficiency display a complete block of B-cell precursor differentiation before the cytoplasmic Igμ-positive pre-B-II cell stage, whereas this blocking of differentiation may be incomplete in patients with hypomorphic RAG mutations [44••].
Artemis Deficiency
Some of the AR T−B−NK+SCID patients harbor no mutations of the RAG1 or RAG2 genes, suggesting that other factors are involved in V(D)J recombination. Indeed, patients with defects of V(D)J recombination can be classified into two distinct groups: one with normal sensitivity to ionizing radiation (RAG1 and RAG2-deficient patients present no radiosensitivity) and the other with higher sensitivity to ionizing radiation (radiosensitive (RS) SCID) [45, 46]. The molecular mechanism for V(D)J recombination is initiated by the creation of DNA double-strand breaks, which are introduced by the lymphoid lineage-specific RAG1 and RAG2 proteins and are repaired by the ubiquitous nonhomologous end-joining (NHEJ) mechanism [47••]. The first genetic etiology of RS SCID was discovered in 2001. Linkage analysis and a candidate gene approach led to the identification of a novel V(D)J recombination/DNA repair factor belonging to the metallo-β lactamase superfamily [48]. This factor is encoded by a gene on chromosome 10p called ARTEMIS (DCLRE1C) (OMIM 602450) [49]. ARTEMIS deficiency accounts for 13 % of all SCID cases [14]. ARTEMIS deficiency is the most common form of RS SCID, and has also been called Athabascan SCID, because it is particularly frequent in the Athabascan population of Native Americans [50, 51]. After allogeneic HSCT, ARTEMIS-deficient patients who had received alkylation therapy in the conditioning regimen were found to have a significantly higher than normal frequency of infections during long-term follow-up, abnormalities of dental development, and endocrine disorders [52••].
Other RS T−B−NK+SCID/NHEJ Pathway Deficiencies
Other molecular defects of genes belonging to the NHEJ pathway have been identified in patients with AR RS T± low B−NK+SCID without ARTEMIS mutation. Several molecules are involved in NHEJ: Ku70, Ku80, and DNA-PKcs (PRKDC gene) together constitute the DNA-dependent protein kinase (DNA-PK), DNA ligase IV (LIG4 gene), X-ray cross-complementation group 4 (XRCC4) and Cernunnos−XRCC4-like factor (XLF) are responsible for the final ligation step [53]. The etiology of RS SCID in humans include deficiencies of PRKDC (OMIM 600899), with the first patient genetically characterized in 2009 [54], LIG4 (OMIM 601837), identified in 2001 [55] and CERNUNNOS/XLF (OMIN 611290), identified in 2006 [56]. LIG4 deficiency is associated with chromosomal instabilities, pancytopenia, microcephaly, developmental and growth delay [57, 58]. Cernunnos-deficient patients also display growth retardation and microcephaly [56]. Genetic defects of the NHEJ pathway render the skin fibroblasts and bone marrow cells of these patients radiosensitive [10] and cause genomic instability, leading to an increase in susceptibility to cancer [59].
T−B+NK+SCIDs
IL-7 Receptor α Deficiency
Some of the patients with AR T−B+NK+SCID have a defect of the alpha subunit of the IL-7 receptor (IL-7Rα) [60]. All these patients have abnormally low T-cell counts, but normal B- and NK-cell counts (T−B+NK+). IL-7Rα is one of the two subunits of IL-7 receptor in addition to γc, and is crucial for IL-7 signaling. More than 20 patients with AR IL-7Rα deficiency (MIM 146661) have been reported [3, 10, 60, 61]. By contrast to the findings for patients with γc and JAK3 deficiencies, NK-cell development and activity appear to be normal in IL-7Rα-deficient patients. The characteristics of this disorder indicate that defects of IL-7/IL-7R signaling are responsible for the lack of T cells in γc- and JAK3-deficient patients. Patients with IL-7Rα deficiency and those with NK−SCID have similar clinical phenotypes, indicating that NK cells cannot compensate for the T-cell deficiency. IL-7Rα deficiency accounts for less than 10 % of all SCID cases [3, 14, 27].
Pre TCR and TCR Complex Deficiencies
Immunological reports of AR T−B+NK+SCID patients with CD3 expression deficiency were first published in 1986 and 1988 [62, 63]. The TCR is a heterodimer consisting of four CD3 chains: the γ, δ, ε, and ζ chains. The CD3G, CD3D, and CD3E genes map to chromosome 11q23 and CD3Z maps to chromosome 1. The first patients with CD3 deficiency to be described were characterized genetically in 1992, with the identification of homozygous mutations of the CD3G gene (OMIN 186740) [64]. AR CD3δ deficiency (OMIM 186790) was subsequently reported, in 2003, in a large Canadian kindred [65]. The first case of AR partial CD3ε deficiency (OMIM 186830) was reported in 1993 [66], and the first case of AR complete CD3ε deficiency was described in 2004 [67]. The first case of AR CD3ζ deficiency (OMIM 186780) was reported in 2006 in a patient with a homozygous germline mutation of CD3Z (CD247) and somatic mutations partially correcting the CD3ζ deficiency [68]. Up to 30 patients with CD3 deficiencies have been reported to date [10]. Patients with CD3δ deficiency, complete CD3ε deficiency, and CD3ζ deficiency have a clinical SCID phenotype, with susceptibility to infection and a complete absence of T-cell function. They account for 3 % of SCID cases [10, 68–70]. Patients with a homozygous mutation of the CD3δ gene have a profound deficiency of mature circulating CD3 T cells, no CD4 or CD8 T cells, and a total absence of γ/δ T cells [10, 65]. Complete CD3ε deficiency has been shown to result in a complete absence of mature TCR α/β and γ/δ cells [69]. Patients with CD3 deficiencies have either normal or higher than normal numbers of B cells and normal numbers of NK cells. Their lymphocytes do not respond to mitogens [65]. Hypomorphic mutations of CD3Ε and mutations of CD3G, result in a partial arrest of T-cell maturation and, therefore, CID, but not an immunological phenotype of SCID with a mild decrease of T-cell activation in assays in vitro [69].
Another rare AR cause of human SCID, with a defect in the expression of CD45 at the cell surface of T cells, was reported in 1997 [71]. Compound heterozygous mutations of a gene encoding protein tyrosine phosphatase, receptor type, C (PTPRC gene) (OMIM 151460) were then identified in 2000, and a homozygous mutation was identified in one unrelated patient in 2001 [72, 73]. PTPRC is also known as the CD45 antigen. This hematopoietic cell-specific transmembrane protein tyrosine phosphatase regulates the Src kinases required for T- and B-cell antigen receptor signal transduction [74]. CD45-deficient patients have been reported to display signs of SCID, with very small numbers of T cells, but high numbers of B cells and normal NK-cell counts. The T cells of these patients fail to respond to mitogens. In 2011, patients with TCRα deficiency (OMIM 186880) were reported in two kindreds affected by CID; the affected patients displayed chronic viral infection, candidiasis, and autoimmunity [75]. Genetic linkage studies mapped the disorder to chromosomal region 14q11.2, and a homozygous mutation of the TRAC gene, encoding the α chain of the TCR was identified [75]. T cells from patients displayed profound impairment of the surface expression of the TCRα/β complex. In patients with T− or low B+NK+ immunological phenotypes, a careful study of TCR CD3 and CD45 molecule expression on residual T cells should be performed by flow cytometry before any genetic studies are undertaken.
PNP Deficiency
This last etiology of AR T−B+NK+SCID was identified by Giblett et al. in 1975, with the demonstration of an absence of the purine nucleoside phosphorylase (PNP) enzyme in affected patients [76]. The gene encoding PNP was mapped to chromosome 14q13.1 by somatic cell hybridization techniques and cloned [77, 78]. The first homozygous mutation of this gene was reported in 1987 [79]. Over 25 deleterious mutations have been identified in 60 families affected by PNP deficiency (OMIM 164050) [10]. PNP deficiency is caused by mutations in this gene, resulting in a marked accumulation of deoxyguanosine triphosphate (dGTP) with undetectable PNP enzyme activity in cells [10]. This dGTP accumulation inhibits the growth of T cells. PNP-deficient patients display T lymphopenia, but they have some residual T cells, accounting for their susceptibility to opportunistic infections being lower than that of patients with other forms of SCID [10]. These patients display a profound T-cell deficiency, with T-cell counts decreasing over time [80]. One third of patients develop autoimmune disease (autoimmune cytopenia and systemic lupus erythematosus). More than half the known PNP-deficient patients also display neurological dysfunctions [80, 81], from spasticity to developmental delay. In patients with T−B+NK+SCID and neurological signs, PNP enzyme activity should be assessed in whole-blood cells if the patients have had a blood transfusion, or in leukocytes, before any genetic tests are carried out.
SCID with Athymia
FOXN1 Deficiency
AR severe T-cell immunodeficiency (T−B+NK+) (OMIM 300400), associated with congenital alopecia of the scalp, eyebrows, and eyelashes, and referred to as Nude/SCID (OMIM 601705), was clinically described in humans in 1996, in two Italian sisters [82]. This phenotype is the human equivalent of the murine phenotype reported by Flanagan in 1966 [83]. The genetic etiology of nude mice was identified in 1994. The causal gene encodes a member of the winged-helix domain family of transcription factors [84]. Initially known as winged-helix nude whn, this gene was subsequently renamed forkhead box n1 (FOXN1) [85]. FOXN1 is tissue-specific, being restricted to the thymus and skin [85]. In 1999, a homozygous R255X nonsense mutation of the FOXN1 gene was identified in the two patients with the Nude/SCID phenotype [86]. Two other unrelated patients were reported in 2011: a Portuguese patient with a homozygous R255X nonsense mutation and a French patient with a homozygous R320W missense mutation [87•]. The absence of the FOXN1 transcription factor results, both in mice and humans, in congenital athymia and hairlessness. This immune defect represents the first example of a SCID phenotype not primarily related to an abnormality intrinsic to hematopoietic cell, instead resulting from a particular change in thymic epithelial cells [88, 89]. In patients with T−B+NK+SCID with alopecia, the FOXN1 gene should to be sequenced.
Complete DiGeorge Syndrome
DiGeorge syndrome (DGS) was first described in the 1960s, in patients with T-cell deficiency (due to thymic hypoplasia), hypoparathyroidism, conotruncal heart defects, and facial dysmorphia [90]. In the 1980s, it was recognized that deletions of the long arm of chromosome 22 at position q.11 were most commonly associated with DGS (OMIM 188400) [91, 92]. DGS has a heterogeneous clinical phenotype, with variable expression of the different clinical features, including the immunodeficiency. DGS may be associated with anything from complete T-cell deficiency (T−B+NK+, complete DGS) to normal T-cell numbers. The T-cell immunodeficiency may be incomplete (T low B+NK+), in which case the term partial DGS is used. The TBX1 gene, which is located on chromosome 22q11, is involved in the thymus development impairment observed in DGS patients [93]. TBX1 is a T-box gene with an important role in regulating the expression of transcription factors. Studies of patients with complete DGS have provided insight into thymus development and the mechanisms of thymopoiesis required to generate broad T cell-mediated immunity [93]. In SCID T−B+NK+ patients with cardiac malformations, hypocalcemia, and dysmorphia, fluorescence in situ hybridization should be carried out to identify small deletions of chromosome 22q11.2.
Patients with a Clinical SCID Phenotype Without T-cell Lymphopenia
Several genetic defects have recently been identified in patients with severe recurrent infections, including opportunistic infections in particular, and in some patients with autoimmunity and lymphoproliferative disorders [10]. A first group of patients with severe recurrent infections have Ca2+ release-activated Ca2+ (CRAC) channelopathies, which were first described in 1995 [94]. These disorders are caused by AR deficiencies of ORAI1 (OMIN 610277) and STIM1 (OMIM 605921), which were identified in 2006 and 2009, respectively [95, 96]. Mutations of the ORAI1 and STIM1 genes in human patients are associated with a unique clinical phenotype characterized by severe immunodeficiency, muscular hypotonia, and anhidrotic ectodermal dysplasia [97]. The immunodeficiency in these patients is caused principally by a severe defect of T-cell activation, rather than of T-cell development [97]. The second group of patients have genetic defects impairing components of the TCR cell signaling pathway. AR ZAP70 deficiency (OMIM 269840) was first described in 1994 and LCK deficiency (OMIM 153390) was reported in 2012 [98–100]. ZAP70-deficient patients have profound T CD8 lymphopenia and LCK-deficient patients display profound T CD4 lymphopenia. Patients with ZAP70 or LCK deficiency have a major T-cell dysfunction, as their T cells are unable to respond to mitogens. The third group of patients has genetic defects impairing T- and B-cell activation and the NF-κB signaling pathway. These defects include the recently identified (in 2013) AR CARD11 deficiency (OMIM 615206), AR MALT1 deficiency (OMIM 604860), and AR IKK2 deficiency (OMIM 603258) [101, 102, 103••, 104]. CARD11 belongs to the CARD protein family, the members of which carry a characteristic caspase-associated recruitment domain. This protein forms a complex with BCL10 and MALT1. This complex forms an essential molecular link between the triggering of cell-surface antigen receptors and nuclear factor-kappa B activation [105]. AR CARD11 deficiency is characterized by hypogammaglobulinemia and a defect of memory B and T cells, and patients have no regulatory T cells or NKT cells [101, 102]. AR MALT1 deficiency is characterized by normal T- and B-cell counts, but low levels of T-cell proliferation [104]. I-kappa-B kinase 2 (IKK2, also known as ΙΚΚβ) is encoded by the IKBKB gene and is a component of the NF-κB pathway. IKK2 deficiency leads to hypogammaglobulinemia or agammaglobulinemia. The B and T cells of patients are almost exclusively of the naive phenotype, and regulatory T cells and Tγ/δ cells are absent [103••]. In cases of a clinical SCID phenotype, extensive immunophenotyping of naive and memory T and B cells, and assays of T-cell proliferation in vitro in response to mitogens (PHA, anti-CD3, and PMA+ionomcyn) should be carried out before genetic analysis.
Conclusion
In conclusion, inherited disorders of SCID can be classified into six groups (Table 1) on the basis of immunophenotyping and functional T-cell activation assays. These disorders are associated with mutations of a total of 27 genes. The first mutations were identified in 1985 (ADA). The number of recognized SCID disorders is expanding: in the last 10 years, ten new disease-causing genes have been identified. In pediatric practice, SCID should be suspected in the following situations: (i) children with multiple infections in the first year of life, (ii) children with recurrent respiratory infections, warts, neutropenia, and hypogammaglobulinemia, (iii) a lack of T cells and/or poorly functional T cells. Immunological studies are required before undertaking genetic analyses of SCID patients. Molecular characterization of these patients is an important issue for genetic counseling and the choice of the most appropriate treatment. Several countries are now developing newborn screening programs for the early identification of patients with SCID disorders. However, newborn screening based on TREC detection can only identify patients with few or no naive T cells, and patients with defects of T-cell activation will be missed. A careful clinical and immunological evaluation of patients presenting warning signs for SCID thus remains the cornerstone for diagnosis.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Chapel H, Geha R, Rosen F. Primary immunodeficiency diseases: an update. Clin Exp Immunol. 2003;132:9–15.
•• Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, Franco JL, Gaspar HB, Holland SM, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162. A comprehensive review on classification of the different molecular defects responsible of primary immunodeficiency.
Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625–55.
Glanzmann E, Riniker P. Essential lymphocytophthisis; new clinical aspect of infant pathology. Ann Paediatr. 1950;175:1–32.
Hitzig WH, Biro Z, Bosch H, Huser HJ. Agammaglobulinemia & alymphocytosis with atrophy of lymphatic tissue. Helvet Paediatr Acta. 1958;13:551–85.
Fischer A. Severe combined immunodeficiencies (SCID). Clin Exp Immunol. 2000;122:143–9.
• Puck JM. The case for newborn screening for severe combined immunodeficiency and related disorders. Ann N Y Acad Sci. 2011;1246:108–17. A comprehensive review about newborn screening for detecting children with severe combined immunodeficiency.
Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. 1972;2:1067–9.
Valerio D, McIvor RS, Williams SR, Duyvesteyn MG, van Ormondt H, van der Eb AJ, Martin DW Jr. Cloning of human adenosine deaminase cDNA and expression in mouse cells. Gene. 1984;31:147–53.
Ochs HD, Smith CIE, Puck J. Primary immunodeficiencies: a molecular and genetic approach. 3rd ed. New York: Oxford University Press; 2013.
Bonthron DT, Markham AF, Ginsburg D, Orkin SH. Identification of a point mutation in the adenosine deaminase gene responsible for immunodeficiency. J Clin Invest. 1985;76:894–7.
Valerio D, Dekker BM, Duyvesteyn MG, van der Voorn L, Berkvens TM, van Ormondt H, van der Eb AJ. One adenosine deaminase allele in a patient with severe combined immunodeficiency contains a point mutation abolishing enzyme activity. EMBO J. 1986;5:113–9.
Shovlin CL, Simmonds HA, Fairbanks LD, Deacock SJ, Hughes JM, Lechler RI, Webster AD, Sun XM, Webb JC, Soutar AK. Adult onset immunodeficiency caused by inherited adenosine deaminase deficiency. J Immunol. 1994;153:2331–9.
Neven B, Leroy S, Decaluwe H, Le Deist F, Picard C, Moshous D, Mahlaoui N, Debre M, Casanova JL, Dal Cortivo L, et al. Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency. Blood. 2009;113:4114–24.
de VO, Seynhaeve V. Reticular dysgenesia. Lancet. 1959;2:1123–5.
Ownby DR, Pizzo S, Blackmon L, Gall SA, Buckley RH. Severe combined immunodeficiency with leukopenia (reticular dysgenesis) in siblings: immunologic and histopathologic findings. J Pediatr. 1976;89:382–7.
Haas RJ, Niethammer D, Goldmann SF, Heit W, Bienzle U, Kleihauer E. Congenital immunodeficiency and agranulocytosis (reticular dysgenesia). Acta Paediatr Scand. 1977;66:279–83.
Pannicke U, Honig M, Hess I, Friesen C, Holzmann K, Rump EM, Barth TF, Rojewski MT, Schulz A, Boehm T, et al. Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet. 2009;41:101–5.
Lagresle-Peyrou C, Six EM, Picard C, Rieux-Laucat F, Michel V, Ditadi A, de Demerens Chappedelaine C, Morillon E, Valensi F, Simon-Stoos KL, et al. Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet. 2009;41:106–11.
de Saint Basile G, Arveiler B, Oberle I, Malcolm S, Levinsky RJ, Lau YL, Hofker M, Debre M, Fischer A, Griscelli C, et al. Close linkage of the locus for X chromosome-linked severe combined immunodeficiency to polymorphic DNA markers in Xq11-q13. Proc Natl Acad Sci USA. 1987;84:7576–9.
Puck JM, Deschenes SM, Porter JC, Dutra AS, Brown CJ, Willard HF, Henthorn PS. The interleukin-2 receptor gamma chain maps to Xq13.1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum Mol Genet. 1993;2:1099–104.
Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, McBride OW, Leonard WJ. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993;73:147–57.
Asao H, Okuyama C, Kumaki S, Ishii N, Tsuchiya S, Foster D, Sugamura K. Cutting edge: the common gamma-chain is an indispensable subunit of the IL-21 receptor complex. J Immunol. 2001;167:1–5.
Puck JM. IL2RGbase: a database of gamma c-chain defects causing human X-SCID. Immunol Today. 1996;17:507–11.
Niemela JE, Puck JM, Fischer RE, Fleisher TA, Hsu AP. Efficient detection of thirty-seven new IL2RG mutations in human X-linked severe combined immunodeficiency. Clin Immunol. 2000;95:33–8.
Stephan JL, Vlekova V, Le Deist F, Blanche S, Donadieu J, De Saint-Basile G, Durandy A, Griscelli C, Fischer A. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr. 1993;123:564–72.
Buckley RH, Schiff RI, Schiff SE, Markert ML, Williams LW, Harville TO, Roberts JL, Puck JM. Human severe combined immunodeficiency: genetic, phenotypic, and functional diversity in one hundred eight infants. J Pediatr. 1997;130:378–87.
Laffort C, Le Deist F, Favre M, Caillat-Zucman S, Radford-Weiss I, Debre M, Fraitag S, Blanche S, Cavazzana-Calvo M, de Saint Basile G, et al. Severe cutaneous papillomavirus disease after haemopoietic stem-cell transplantation in patients with severe combined immune deficiency caused by common gammac cytokine receptor subunit or JAK-3 deficiency. Lancet. 2004;363:2051–4.
Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, Migone TS, Noguchi M, Markert ML, Buckley RH, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995;270:797–800.
Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O’Shea JJ, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature. 1995;377:65–8.
Candotti F, Oakes SA, Johnston JA, Giliani S, Schumacher RF, Mella P, Fiorini M, Ugazio AG, Badolato R, Notarangelo LD, et al. Structural and functional basis for JAK3-deficient severe combined immunodeficiency. Blood. 1997;90:3996–4003.
Schumacher RF, Mella P, Lalatta F, Fiorini M, Giliani S, Villa A, Candotti F, Notarangelo LD. Prenatal diagnosis of JAK3 deficient SCID. Prenat Diagn. 1999;19:653–6.
Buckley RH, Schiff SE, Schiff RI, Markert L, Williams LW, Roberts JL, Myers LA, Ward FE. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med. 1999;340:508–16.
Vihinen M, Villa A, Mella P, Schumacher RF, Savoldi G, O’Shea JJ, Candotti F, Notarangelo LD. Molecular modeling of the Jak3 kinase domains and structural basis for severe combined immunodeficiency. Clin Immunol. 2000;96:108–18.
Frucht DM, Gadina M, Jagadeesh GJ, Aksentijevich I, Takada K, Bleesing JJ, Nelson J, Muul LM, Perham G, Morgan G, et al. Unexpected and variable phenotypes in a family with JAK3 deficiency. Genes Immun. 2001;2:422–32.
Notarangelo LD, Mella P, Jones A, de Saint Basile G, Savoldi G, Cranston T, Vihinen M, Schumacher RF. Mutations in severe combined immune deficiency (SCID) due to JAK3 deficiency. Hum Mutat. 2001;18:255–63.
Roberts JL, Lengi A, Brown SM, Chen M, Zhou YJ, O’Shea JJ, Buckley RH. Janus kinase 3 (JAK3) deficiency: clinical, immunologic, and molecular analyses of 10 patients and outcomes of stem cell transplantation. Blood. 2004;103:2009–18.
Ichihara Y, Matsuoka H, Tsuge I, Okada J, Torii S, Yasui H, Kurosawa Y. Abnormalities in DNA rearrangements of immunoglobulin gene loci in precursor B cells derived from X-linked agammaglobulinemia patient and a severe combined immunodeficiency patient. Immunogenetics. 1988;27:330–7.
Schwarz K, Hansen-Hagge TE, Knobloch C, Friedrich W, Kleihauer E, Bartram CR. Severe combined immunodeficiency (SCID) in man: B cell-negative (B-) SCID patients exhibit an irregular recombination pattern at the JH locus. J Exp Med. 1991;174:1039–48.
Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–48.
van Gent DC, Ramsden DA, Gellert M. The RAG1 and RAG2 proteins establish the 12/23 rule in V(D)J recombination. Cell. 1996;85:107–13.
Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, Friedrich W, Seger RA, Hansen-Hagge TE, Desiderio S, et al. RAG mutations in human B cell-negative SCID. Science. 1996;274:97–9.
Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, Schneider DT, Manfras B, Pannicke U, Willemze R, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. 2008;358:2030–8.
•• van der Burg M, Gennery AR. Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur J Pediatr. 2011;170:561–71. An excellent review about human severe combined immunodeficiency. This manuscript describes the clinical and immunological features of each molecular defect.
Cavazzana-Calvo M, Le Deist F, De Saint Basile G, Papadopoulo D, De Villartay JP, Fischer A. Increased radiosensitivity of granulocyte macrophage colony-forming units and skin fibroblasts in human autosomal recessive severe combined immunodeficiency. J Clin Invest. 1993;91:1214–8.
Nicolas N, Moshous D, Cavazzana-Calvo M, Papadopoulo D, de Chasseval R, Le Deist F, Fischer A, de Villartay JP. A human severe combined immunodeficiency (SCID) condition with increased sensitivity to ionizing radiations and impaired V(D)J rearrangements defines a new DNA recombination/repair deficiency. J Exp Med. 1998;188:627–34.
•• Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet. 2011;45:167–202. An excellent and comprehensive review on VDJ recombination genetic defects.
Moshous D, Li L, Chasseval R, Philippe N, Jabado N, Cowan MJ, Fischer A, de Villartay JP. A new gene involved in DNA double-strand break repair and V(D)J recombination is located on human chromosome 10p. Hum Mol Genet. 2000;9:583–8.
Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–86.
Dvorak CC, Cowan MJ. Radiosensitive severe combined immunodeficiency disease. Immunol Allergy Clin N Am. 2010;30:125–42.
Li L, Moshous D, Zhou Y, Wang J, Xie G, Salido E, Hu D, de Villartay JP, Cowan MJ. A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans. J Immunol. 2002;168:6323–9.
•• Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, Schwarz K, Schulz AS, Hoenig M, Sparber-Sauer M, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood 2014;123:281–9. An excellent manuscript on patients with T-B-NK+SCID disorders. This manuscript describes the clinical and immunological features of each molecular defect, and discusses the therapeutic approaches.
Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211.
van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, Mari PO, Tezcan I, Chen DJ, Zdzienicka MZ, et al. A DNA-PKcs mutation in a radiosensitive T-B-SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009;119:91–8.
O’Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, Hirsch B, Gennery A, Palmer SE, Seidel J, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8:1175–85.
Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, Plebani A, Stephan JL, Hufnagel M, le Deist F, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–99.
van der Burg M, van Veelen LR, Verkaik NS, Wiegant WW, Hartwig NG, Barendregt BH, Brugmans L, Raams A, Jaspers NG, Zdzienicka MZ, et al. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. J Clin Invest. 2006;116:137–45.
Buck D, Moshous D, de Chasseval R, Ma Y, le Deist F, Cavazzana-Calvo M, Fischer A, Casanova JL, Lieber MR, de Villartay JP. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur J Immunol. 2006;36:224–35.
Kasparek TR, Humphrey TC. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin Cell Dev Biol. 2011;22:886–97.
Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat Genet. 1998;20:394–7.
Roifman CM, Zhang J, Chitayat D, Sharfe N. A partial deficiency of interleukin-7R alpha is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Blood. 2000;96:2803–7.
Regueiro JR, Arnaiz-Villena A, de Ortiz Landazuri M, Martin Villa JM, Vicario JL, Pascual-Ruiz V, Guerra-Garcia F, Alcami J, Lopez-Botet M, Manzanares J. Familial defect of CD3 (T3) expression by T cells associated with rare gut epithelial cell autoantibodies. Lancet. 1986;1:1274–5.
Alarcon B, Regueiro JR, Arnaiz-Villena A, Terhorst C. Familial defect in the surface expression of the T-cell receptor-CD3 complex. N Engl J Med. 1988;319:1203–8.
Arnaiz-Villena A, Timon M, Corell A, Perez-Aciego P, Martin-Villa JM, Regueiro JR. Brief report: primary immunodeficiency caused by mutations in the gene encoding the CD3-gamma subunit of the T-lymphocyte receptor. N Engl J Med. 1992;327:529–33.
Dadi HK, Simon AJ, Roifman CM. Effect of CD3delta deficiency on maturation of alpha/beta and gamma/delta T-cell lineages in severe combined immunodeficiency. N Engl J Med. 2003;349:1821–8.
Soudais C, de Villartay JP, Le Deist F, Fischer A, Lisowska-Grospierre B. Independent mutations of the human CD3-epsilon gene resulting in a T cell receptor/CD3 complex immunodeficiency. Nat Genet. 1993;3:77–81.
de Saint Basile G, Geissmann F, Flori E, Uring-Lambert B, Soudais C, Cavazzana-Calvo M, Durandy A, Jabado N, Fischer A, Le Deist F. Severe combined immunodeficiency caused by deficiency in either the delta or the epsilon subunit of CD3. J Clin Invest. 2004;114:1512–7.
Rieux-Laucat F, Hivroz C, Lim A, Mateo V, Pellier I, Selz F, Fischer A, Le Deist F. Inherited and somatic CD3zeta mutations in a patient with T-cell deficiency. N Engl J Med. 2006;354:1913–21.
Fischer A, de Saint Basile G, Le Deist F. CD3 deficiencies. Curr Opin Allergy Clin Immunol. 2005;5:491–5.
Roberts JL, Lauritsen JP, Cooney M, Parrott RE, Sajaroff EO, Win CM, Keller MD, Carpenter JH, Carabana J, Krangel MS, et al. T-B+NK+ severe combined immunodeficiency caused by complete deficiency of the CD3zeta subunit of the T-cell antigen receptor complex. Blood. 2007;109:3198–206.
Cale CM, Klein NJ, Novelli V, Veys P, Jones AM, Morgan G. Severe combined immunodeficiency with abnormalities in expression of the common leucocyte antigen, CD45. Arch Dis Child. 1997;76:163–4.
Kung C, Pingel JT, Heikinheimo M, Klemola T, Varkila K, Yoo LI, Vuopala K, Poyhonen M, Uhari M, Rogers M, et al. Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nat Med. 2000;6:343–5.
Tchilian EZ, Wallace DL, Wells RS, Flower DR, Morgan G, Beverley PC. A deletion in the gene encoding the CD45 antigen in a patient with SCID. J Immunol. 2001;166:1308–13.
Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21:107–37.
Morgan NV, Goddard S, Cardno TS, McDonald D, Rahman F, Barge D, Ciupek A, Straatman-Iwanowska A, Pasha S, Guckian M, et al. Mutation in the TCRalpha subunit constant gene (TRAC) leads to a human immunodeficiency disorder characterized by a lack of TCRalphabeta+T cells. J Clin Invest. 2011;121:695–702.
Giblett ER, Ammann AJ, Wara DW, Sandman R, Diamond LK. Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet. 1975;1:1010–3.
Ricciuti F, Ruddle FH. Assignment of nucleoside phosphorylase to D-14 and localization of X-linked loci in man by somatic cell genetics. Nature. 1973;241:180–2.
Williams SR, Goddard JM, Martin DW Jr. Human purine nucleoside phosphorylase cDNA sequence and genomic clone characterization. Nucleic Acids Res. 1984;12:5779–87.
Williams SR, Gekeler V, McIvor RS, Martin DW Jr. A human purine nucleoside phosphorylase deficiency caused by a single base change. J Biol Chem. 1987;262:2332–8.
Markert ML. Purine nucleoside phosphorylase deficiency. Immunodefic Rev. 1991;3:45–81.
Simmonds HA, Fairbanks LD, Morris GS, Morgan G, Watson AR, Timms P, Singh B. Central nervous system dysfunction and erythrocyte guanosine triphosphate depletion in purine nucleoside phosphorylase deficiency. Arch Dis Child. 1987;62:385–91.
Pignata C, Fiore M, Guzzetta V, Castaldo A, Sebastio G, Porta F, Guarino A. Congenital alopecia and nail dystrophy associated with severe functional T-cell immunodeficiency in two sibs. Am J Med Genet. 1996;65:167–70.
Flanagan SP. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet Res. 1966;8:295–309.
Nehls M, Pfeifer D, Schorpp M, Hedrich H, Boehm T. New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature. 1994;372:103–7.
Schorpp M, Hofmann M, Dear TN, Boehm T. Characterization of mouse and human nude genes. Immunogenetics. 1997;46:509–15.
Frank J, Pignata C, Panteleyev AA, Prowse DM, Baden H, Weiner L, Gaetaniello L, Ahmad W, Pozzi N, Cserhalmi-Friedman PB, et al. Exposing the human nude phenotype. Nature. 1999;398:473–4.
• Markert ML, Marques JG, Neven B, Devlin BH, McCarthy EA, Chinn IK, Albuquerque AS, Silva SL, Pignata C, de Saint Basile G, et al. First use of thymus transplantation therapy for FOXN1 deficiency (nude/SCID): a report of 2 cases. Blood. 2011;117:688–96. This paper reports the first thymic engraftment in patients with FOXN1 deficiency.
Pignata C. A lesson for unraveling complex aspects of novel immunodeficiencies from the human equivalent of the nude/SCID phenotype. J Hematother Stem Cell Res. 2002;11:409–14.
Palamaro L, Romano R, Fusco A, Giardino G, Gallo V, Pignata C. FOXN1 in organ development and human diseases. Int Rev Immunol. 2014;33:83–93.
Kirkpatrick JA Jr, DiGeorge AM. Congenital absence of the thymus. The American journal of roentgenology, radium therapy, and nuclear medicine. 1968;103:32–7.
de la Chapelle A, Herva R, Koivisto M, Aula P. A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet. 1981;57:253–6.
Kelley RI, Zackai EH, Emanuel BS, Kistenmacher M, Greenberg F, Punnett HH. The association of the DiGeorge anomalad with partial monosomy of chromosome 22. J Pediatr. 1982;101:197–200.
Hollander G, Gill J, Zuklys S, Iwanami N, Liu C, Takahama Y. Cellular and molecular events during early thymus development. Immunol Rev. 2006;209:28–46.
Le Deist F, Hivroz C, Partiseti M, Thomas C, Buc HA, Oleastro M, Belohradsky B, Choquet D, Fischer A. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood. 1995;85:1053–62.
Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–85.
Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux-Laucat F, Rechavi G, Rao A, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360:1971–80.
Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev. 2009;231:189–209.
Chan AC, Kadlecek TA, Elder ME, Filipovich AH, Kuo WL, Iwashima M, Parslow TG, Weiss A. ZAP-70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science. 1994;264:1599–601.
Elder ME, Lin D, Clever J, Chan AC, Hope TJ, Weiss A, Parslow TG. Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science. 1994;264:1596–9.
Hauck F, Randriamampita C, Martin E, Gerart S, Lambert N, Lim A, Soulier J, Maciorowski Z, Touzot F, Moshous D, et al. Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency. J Allergy Clin Immunol. 2012;130(1144–1152):e1111.
Stepensky P, Keller B, Buchta M, Kienzler AK, Elpeleg O, Somech R, Cohen S, Shachar I, Miosge LA, Schlesier M, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol. 2013;131:477–85.
Greil J, Rausch T, Giese T, Bandapalli OR, Daniel V, Bekeredjian-Ding I, Stutz AM, Drees C, Roth S, Ruland J, et al. Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol. 2013;131(1376–1383):e1373.
•• Pannicke U, Baumann B, Fuchs S, Henneke P, Rensing-Ehl A, Rizzi M, Janda A, Hese K, Schlesier M, Holzmann K, et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N Engl J Med. 2013;369:2504–14. This paper reports the first identification of mutation in the IKBKB gene in patients.
Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, Chouery E, Mikhael R, Gorka O, Gewies A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol. 2013;132:151–8.
Turvey SE, Durandy A, Fischer A, Fung SY, Geha RS, Gewies A, Giese T, Greil J, Keller B, McKinnon ML, et al. The CARD11-BCL10-MALT1 (CBM) signalosome complex: stepping into the limelight of human primary immunodeficiency. J Allergy Clin Immunol. 2014;134:276–84.
Acknowledgments
We would like to thank all the members of our laboratory and unit for helpful discussions, and critical reading of the text, and our collaborators worldwide for their trust and patience.
Disclosure
Capucine Picard, Despina Moshous, and Alain Fischer declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Immunology.
Rights and permissions
About this article

Cite this article
Picard, C., Moshous, D. & Fischer, A. The Genetic and Molecular Basis of Severe Combined Immunodeficiency. Curr Pediatr Rep 3, 22–33 (2015). https://doi.org/10.1007/s40124-014-0070-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-014-0070-8