
Abstract
In this study, the electrical and structural parameters of pristine and 1Ca- and 2Ca-doped beryllium oxide nanotubes (BeONTs) before and after N2O adsorption are studied using density function theory (DFT). In the first step, we selected 15 models for the adsorption of N2O gas on the exterior and interior surfaces of nanotube and then the considered models are optimized using the B3LYP/6-31G(d, p) level of theory. The results indicate that the adsorption processes in all the models are physisorption and are endothermic. A strong interaction between N2O and 1Ca-, 2Ca-doped BeONTs increases the conductivity of nanotube, which acts a good candidate for make sensor for N2O gas. The ESP analysis shows that the nanotube is relatively electron rich in N2O/BeONTs complex, and the N2O is relatively electron poor. With 1Ca and 2Ca doping, stabilization energy (E 2) and charge density of three oxygen atoms around the dopant decrease and the dipole moment of nanotube increases significantly from original values.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nitrous oxide (N2O) gas is a colorless, non-flammable gas, with a slightly sweet odor and taste. It is known as “laughing gas” due to the euphoric effects of inhaling it [1]. N2O gas has been generated as a byproduct of nitric and adipic acids [2–4]. It is used in surgery, dentistry, as a powerful oxidizer in rocket propellants, in motor racing, and as a good solvent for many organic compounds [5–7]. N2O gas extremely reacts with ozone in the stratosphere and acts as a regulator of stratospheric ozone and it is known also as air pollutant and a major greenhouse gas [8–10]. Therefore, extensive research has been carried out to control and monitor N2O gas to overcome the human health and environmental concerns caused by it. Recent investigations indicated that CuO, BeO, In2O3, SnO2, WO3, and ZnCdO can be employed for monitoring and adsorbing N2O gas [10, 11]. The interaction of N2O gas with alkaline earth oxides [12–17], TiO2 [18], molecular zeolite and metals [19–26], isolated Cu+ ion [27, 28], BNNTs surfaces, Al-doped (6,0) zigzag SWCNTs, AlNNTs, and AlPNTs, Co-doped MgONTs, BeONTs, and ZnONTs have been extensively investigated in many fields [29–34]. After the discovery of carbon nanotube and studying the structural parameters and applications of it, many efforts have been focused to find other nanotubes. One of them was beryllium oxide nanotube (BeONTs). After the discovery of BeONTs by Continenza et al. in 1990 [35], Baumeier et al. and Wu et al. demonstrated that the fluorinated and hydrogenated BeO nanosheets behave as semiconductors [36–38]. The other computational studies showed that B, C, and N dopant impurity atoms decreased the energy gap of BeONTs and effectively improved the electronic structure, optical properties, magnetism, and other applications of nanotube [39–41]. In the recent years, interaction and adsorption of H2, CO2, H2S, and N2O gas with pristine BeONTs were investigated, and these results indicated BeONTs as a good candidate for adsorbing gas and making a gas sensor [40–43]. For this aim, in the current research, the effects of 1Ca- and 2Ca-doped BeONTs on the adsorption of N2O on both exterior and interior surfaces of nanotube are studied using density functional theory and the B3LYP/(d, p) level of theory, in order to reveal some clues for chemical sensor design. These results can provide valuable information about its interaction and reactivity of N2O gas with pristine and Ca-doped BeO nanotube.
Computational methods
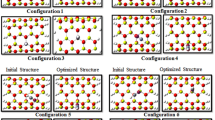
For finding stable adsorption structures, we consider many different configurations and after optimizing all configurations with small basis set, fifteen favorable configurations are selected for this study and are named AI, AII, AIII, BI, BII, BIII, CI, CII, CIII, DI, DII, DIII, EI, EII, and EIII models (see Fig. 1). Here, the A and B models are used to identify the vertical adsorption of N2O gas from the O side and N side on the outer surface of nanotube, respectively, whereas the C and D models are used to show the horizontal adsorption of N2O gas on the forward and backward surfaces of nanotube, respectively. The E model is utilized for the adsorption of N2O gas on the inner surface of nanotube. In all models, indexes I, II, and III are used to depict the pristine and 1Ca- and 2Ca-doped BeONTs, respectively. Moreover, all the models are optimized by B3LYP/6-31G (d, p) basis set using the GAMESS program package [44].

2D views of the optimized structures of N2O adsorption on the surface of pristine and 1Ca- and 2Ca-doped BeONTs (models AI–EIII)
The adsorption energy (E ads) for N2O gas on the surface of the pristine and 1Ca- and 2Ca-doped BeONTs can be computed using the following equation:
where \(E_{{{\text{BeONTs}}{\rm N}_{2} {\rm O}}}\) is the total energy of the complex consisting of N2O gas and BeONTs, while E BeONTs and \(E_{{{\rm N}_{2} {\rm O}}}\) are the total energies of BeONTs and N2O gas, respectively, and BSSE is the basis set superposition error for the adsorption energy. Here, the negative and positive values of the E ads indicate exothermic and endothermic reaction, respectively.
The gap energy (E g) between HOMO and LUMO orbital and Fermi level (E FL) are calculated using the following two equations:
According to Parr et al. [45, 46], the chemical potential (μ) and electronegativity (χ) are defined as follows:
According to Koopmans theorem [47], global hardness (η), the fractional number of electrons transfer (∆N), global softness (S), and electrophilicity index (ω) can be approximated using the following equations:
where E HOMO is the energy of the highest occupied molecular orbital and E LUMO is the energy of the lowest unoccupied molecular orbital of the considered structure. I (−E HOMO) is the ionization potential and A (−E LUMO) is the electron affinity of the molecule.
The chemical shielding isotropic (CSI) and chemical shielding anisotropic (CSA) of the sites of 7Be, 17O nuclei are calculated using the following equations [48–50]:
Results and discussion
Structural geometry of N2O adsorption on the pristine and Ca-doped BeONTs
The 15 optimized models (AI–EIII models) designed for the N2O gas adsorption on the outer and inner surfaces of pristine and 1Ca- and 2Ca-doped BeONTs are depicted in Fig. 1. The length and the diameter of the pristine (4, 4) armchair BeONTs are calculated to be about 9.99 and 6.32 Å, respectively. The optimized geometries including bond lengths and bond angles for all the adsorption models are listed in Table 1. The average bond length and bond angle for the pristine BeONTs are 1.57 Å and 119°, respectively, and it is in agreement with other research studies [39, 40]. The average bond length and bond angle of Be–O and Ca–O in the AII, AIII, BI, BII, CII, CIII, DII, DIII, EII, and EIII are 1.54 and 2.23 Å, respectively (Table 1). In both cases, the Ca dopant atom protrudes a little out of the nanotube surface to occupy more space due to its relatively larger atomic radius than that of Be atom. It is also found that the adsorption persuades little local structural deformation on both the N2O molecule and the BeONTs. The bond length and bond angle of the N2O gas differ slightly between the adsorption models.
The calculated adsorption energies (E ads), dipole moment (µ D), NBO charge transfer of N2O (Qe), and distance between N2O and nanotube (d) for the AI to EIII models are listed in Table 2. The E ads for all models is in the range of 37.73–61.91 kcal/mol; the distance between N2O and nanotube (d) is in the range of 2.42–3.20 Å; and NBO charge transfer of N2O (Qe) for all the models is in the range 0.04–0.08e. The BSSE values for all the models are in the range of 0.002–0.005. The positive values of E ads and the small transferred charge from N2O to the nanotube indicate that the weak adsorption of N2O gas on the surface of BeONTs which is endothermic and also the process is called physical adsorption or physisorption. On the other hand, comparing our results with adsorbing energy of N2O gas on the surface of CNTs [30], AlNNTs [31], and TiO2 [33] show that the adsorption process on the surface of BeONTs is not favorable in thermodynamic approach. This result is in agreement with other research study [34].
HOMO and LUMO orbital energy
In order to study the adsorption properties of N2O gas on the surface of the pristine and 1Ca- and 2Ca-doped BeONTs, the electronic energies, the highest occupied molecular orbital (HOMO), and the lowest unoccupied molecular orbital (LUMO) for all the AI–EIII adsorption modes are calculated, the results of which are shown in Fig. 2. As we can see in the AI–EIII models, HOMO orbitals are uniformly distributed inward of the nanotube and LUMO orbitals are localized around N2O gas. The positive values of NBO charge around N2O gas is also in agreement with the density of LUMO orbital around N2O. To study the chemical activity of the nanotube, the HOMO and LUMO energy, gap energy (E g), and the other quantum descriptors involving the chemical potential (μ), electronegativity (χ), chemical hardness (η), chemical softness (S), and electrophilicity index (ω) are calculated and shown in Table 3 and Table S2. As seen in Table S2, the E g of pristine and 1Ca- and 2Ca-doped BeONTs are 7.12, 4.84, and 4.81 eV, respectively, and it is notable that with 1Ca and 2Ca doping, the gap energy decreases significantly from original value and thereby increasing the conductivity of nanotube. Inspection of calculated results in Table 3 indicates that in all models, the E g values are in the range of 3.74–6.53 eV and slightly lower than original values, as the HOMO and LUMO levels move to higher energies after N2O adsorption.

2D views of the HOMO–LUMO structures of N2O adsorption on the surface of pristine and 1Ca- and 2Ca-doped BeONTs (models AI–EIII)
To facilitate a more detailed study on the change in gap energy, the reduction in percent for E g (%ΔE g) is calculated using the following:
The %ΔE g values in all the adsorption models decrease in the following order: EI > DI > BII > BIII > AII = DII > BI = CI > AIII = DIII > AI > EII > EIII > CIII > CII (see Table 3). Comparison results indicate that the %ΔE g values in CII and CIII models (0.40 and 0.56 %, respectively) are lower than those in the other models, whereas %ΔE g values in DI and EI (22.75 and 23.31 %, respectively) are more than those in the other models.
In order to gain a better understanding about the adsorption process, the density of state (DOS) plots and total charge density around the nanotube are determined from the output of HOMO–LUMO, and the results are shown in Fig. 3. From Fig. 3, it is found that the total charge densities of AI, BI, CI, DI, and EI models are uniformly distributed on the inner layer of the nanotube, but with 1Ca and 2Ca doping in AII, AIII, BII, BIII, CII, CIII, DII, DIII, EII, and EIII models, total charge density gets distributed into the various inner surfaces of the nanotube. Meanwhile, the total electron density and scheme of DOS plots depended on the orientations of N2O gas on the surface of the nanotube. Comparison of the DOS plots exhibits that the electronic properties of nanotube after doping with 1Ca and 2Ca atoms and also N2O adsorption changed significantly. It can be clearly observed that in the AII, BII, CII, DII, EII, AIII, BIII, CIII, DIII and EIII adsorption models one and two picks are generated in the gap region due to 1Ca and 2Ca dopants, and so the gap energy reduced remarkably from original values, thereby increasing the electrical conductivity of nanotube. From these results, it can be proposed that the 1Ca- and 2Ca-doped BeONTs are good candidates for making sensor for N2O gas.

2D views of the DOS plot and total density electron of N2O adsorption on the surface of pristine and 1Ca- and 2Ca-doped BeONTs (models AI–EIII)
To further study the adsorption process, the quantum parameters involving global hardness, the chemical potential, and electronegativity are calculated using Eqs. (3−5) and the results are shown in Table 3 and Fig S4. As is evident from Fig S4, upon adsorbing N2O gas, the global hardness of pristine BeONTs decreased from 3.56 to 3.26 eV and that of 1Ca- and 2Ca-doped BeONTs decreased from 2.42 to 1.95 and 2.35 to 1.99 eV, respectively. This decrease in global hardness leads to decrease in stability and increase in reactivity of the species. On the other hand, the calculated results of all adsorption models show that the electronic chemical potentials and electrophilicity of BeONTs will be decreased.
The positive values of the fractional number of electron transfer (∆N) indicate that when N2O gas is adsorbed on the outer and inner surfaces of the BeONTs, the charge transfer occurred from N2O gas toward nanotube, which suggests that their electronic transport properties could be altered upon adsorptions of N2O. According to thermodynamic approach, the direction of electron flow will occur from higher chemical potential to the lower electronic chemical potential, until the electronic chemical potentials become equal. The chemical potential of the nanotube is lower than N2O gas, and so the electron flow occurs from N2O gas toward the nanotube. The electrophilicity index determines the maximum flow of electron from donor to acceptor species and supplies data connected to structural stability and reactivity of chemical species. It is notable that the electrophilicity index of 1Ca- (4.79 eV) and 2Ca (4.81 eV)-doped BeONTs is more than pristine (2.23 eV) due to the donor electron effect of dopant atoms. On the other hand, when N2O gas adsorbed on the surface of BeONTs in the AI–EIII models the electrophilicity index increased significantly from the original values, indicating that the direction of the charge flow occurred from N2O gas toward the nanotube. Meanwhile, this result emphasizes that the adsorption of N2O gas on the surface of pristine and 1Ca- and Ca-doped BeONTs significantly alters their electronic properties and demonstrates that only a weak interaction exists between them. Here, we also investigated the changes of Fermi energy (E Fermi) for adsorbing N2O gas on the pristine and 1Ca- and 2Ca-doped BeONTs. Inspection of the calculated result in Fig S4 and Tables S2 and S3 reveals that the E Fermi values of nanotube after interaction with N2O gas reduced from −3.94 eV to −4.42, −4.56, −4.56, −4.67, and −4.47 eV in the AI, BI,CI, DI, and EI models, respectively. For 1Ca–doped ones, the E Fermi values of nanotube decreased from −4.81 eV to −5.20, −5.30, and −5.20 eV in the AII, BII, and DII models, respectively. For 2Ca-doped ones, the E Fermi values of nanotube reduced from −4.81 eV to −5.01, −5.11, and −5.01 eV in the AIII, BIII, and DIII models, respectively. It is notable that the E Fermi values of nanotube in the CII, CIII, EII, and EIII increased slightly from the original values. The changes in the Fermi level energy demonstrate that a remarkable number of electrons transfer during the interaction between nanotube and N2O gas; therefore, the electronic structure of the adsorption system changed and the electrical conductance of the material thus is significantly altered.
NMR parameters
The NMR parameters involving the CSI and CAS of 9Be and 17O nuclei are calculated using Eqs. 10 and 11, and the results are shown in Tables S3–S14 and Fig S5. The CSI values of 9Be and 17O nuclei in all AI–EIII models are in the range of 108–112 and 230–253 ppm, respectively. Comparison of the results indicates that with adsorption of N2O gas, the CSI values of nanotube increase significantly from unabsorbed models due to the donor electron effect of N2O gas. To further study the electronic structures of nanotube/N2O complex, the shielding density plots for XX and YY tensors are displayed in Fig S6. The results indicate that in the AI–EIII adsorption models, the electrical charges are distributed continuously around XX direction, whereas the charge density is distributed discontinuously around YY direction due to the electrical effects of N2O gas on the surface of nanotube.
The NMR plots of O and Be atoms for all adsorption models are shown in Fig S7. According to the obtained NMR results, it can be clearly observed that the shielding parameters for O atoms in the [AI and EI] and the [BI and CI] models are in the range of 140–248 and 100–252 ppm, respectively, and for DI model it is in the range of 56–252 ppm. On the other hand, in the AII, DII, and EII models, the shielding parameters are in the range of 132–258 ppm, whereas in the BII and CII models, they are in the range of 100–258 and 88–260 ppm, respectively. And also the shielding parameters in AIII, DIII, and EIII models are in the range of 132–280 ppm, while in BIII and CIII models they are in the range of 100–282 and 84–282 ppm, respectively. Comparing results reveal that with doping 1Ca and 2Ca the shielding parameters values for O atoms increase significantly from original values due to donor electron effect of Ca doped. However, the shielding parameters for Be atoms in all adsorption models (A, B, C, D, and E models) are in the range of 108.5–111 ppm, which slightly vary from original values with Ca doping.
Electrostatic potential maps
To further elucidate the adsorption process, we have calculated the electrostatic potential at selected points on the 0.0002 isodensity surfaces. Electrostatic potential maps are very useful three-dimensional diagrams of molecules. They enable us to visualize the charge distributions of molecules and charge-related properties of molecules. They also allow us to visualize the size and shape of molecules. In organic chemistry, electrostatic potential maps are invaluable in predicting the behavior of complex molecules.
Here, different colors are used to identify different potential of matter. The most negative and positive potentials are colored in red and blue, respectively. Intermediate potentials are assigned colors according to the color spectrum: Red < Orange < Yellow < Green < Blue.
Based on this scheme, one can usually identify red regions of a map as being the most electron-rich regions of a molecule and blue regions of a map as being the most electron-poor regions of a molecule. To accurately analyze the charge distribution of a molecule, a very large quantity of electrostatic potential energy values must be calculated. The best way to convey the data is to visually represent it, as in an electrostatic potential map [51–54]. Electrostatic potential (ESP) maps of all adsorption models (AI–EII) are shown in Fig. 4. Inspections of the results indicate that red regions are found on the surface of nanotube and blue regions are found on the surface of N2O gas. This means that the nanotube is relatively electron rich in this molecule, and the N2O is relatively electron poor.

ESP plots of N2O adsorption on the surface of pristine and 1Ca- and 2Ca-doped BeONTs (models AI–EIII)
NBO analysis
Natural bond orbital (NBO) analysis is an important technique for investigating covalent and hybridization effects in polyatomic wave functions, which is based on local block Eigen vector of the one-particle density matrix. NBO analysis provides information about interaction between both filled and virtual orbital spaces, which in turn could provide information about intra- and intermolecular interactions. For each donor (i) and acceptor (j), the stabilization energy E (2) is estimated from the second-order perturbation approach [55–60] as given below:
where q i is the donor orbital occupancy \(\varepsilon_{i}\) and \(\varepsilon_{j}\) are the diagonal elements, and F(i, j) is the off-diagonal NBO Fock matrix element. These calculations allow us to analyze the probable charge transfers and the intermolecular bond paths [56, 57]. The larger the E (2) value, the more intensive is the interaction, i.e., the more donation tendency from electron donors to electron acceptors and the greater the extent of conjugation in the whole system [58]. In this study, NBO analysis and second-order perturbation theory analysis of the AI–EIII models are presented in Table 4 and Tables S15–S19. The intramolecular interaction is formed by the orbital overlap between σBe62–O52 and σ*Be42–O52 bond orbital, σBe31–O41 → σ*Be41–O41, which results in intramolecular charge transfer causing stabilization of the system. The most interactions in the AI–EIII models occur in the range of 0.67–5.67 kcal/mol. A very strong interaction has been observed due to the electron density transfer from the σO42–Be52 to σ*Be32–O42 with stabilization energy of 6.58 kcal/mol for CI model. This indicates that the strongest charge transfer is responsible for interaction between N2O gas and nanotube in this model. Furthermore, for σBe62–O52 to σ*Be42–O52 in the AIII and EIII models, due to the smaller E (2) of less than about 0.67 kcal/mol, weaker interaction occurred between them. These facts may be the probable reasons behind the relative stability of the axial and equatorial adsorptions of N2O gas on the outer and inner surfaces of nanotube based on energetic data and NBO interpretation. The obtained E (2) values for the interaction between σBe62–O52 and σ*Be42–O52 of the studied AI, BI, CI, DI, and EI models decreased in the following order: AI > BI > CI > EI > DI; and for AII, BII, CII, DII, and EII models the values decreased in the following order: EII > CII > AII = DII > BII; moreover, for AIII, BIII, CIII, DIII, and EIII models, the values decreased in the order as follows: CIII > AIII = EIII = DIII > BIII.
It can be clearly observed that E (2) values in adsorption of N2O gas on the pristine and 1Ca and 2Ca dopants decrease in the following order: pristine (I) > 1Ca-doped (II) > 2Ca-doped.
We found that with 1Ca and 2Ca atom doping, the charge transfer between σBe62–O52 and σ*Be42–O52 decreases significantly from original values; on the other hand, the orientation of N2O adsorbed significantly influences the charge transfer. Inspection of the dipole moment (µ D) results at Table 2 reveal that E (2) has the linear relation with the dipole moment inversion. On the other hand with doping one and two Ca atoms the dipole moment of systems increase significantly from original values. Therefore, dopants 1Ca and 2Ca atoms bollix the structures of nanotube thus increasing the dipole moment and decreasing the charge transfer in the σBe62–O52 and σ*Be42–O52 bond orbital. In addition, from Table 2, it can be inferred that with 1Ca and 2Ca atom doping, the NBO charge on the oxygen atoms decreases significantly due to the donor electron effect of the dopant atom, and so the Be–O bonds are highly polarized and are responsible for the large shift of charge from metal to its neighbors. It is found that there is small back-bonding interaction for Ca which account for the fact that it has large charge distribution and smaller covalent contribution, this also supports the high adsorption energy of Ca.
Conclusions
In this research, the electrical and structural parameters of pristine and 1Ca- and 2Ca-doped BeONTs, both on the exterior and interior surfaces, before and after N2O adsorption are investigated using density function theory. The computational results established that the adsorption process in all models is endothermic. The decrease in percentage of gap energy (%ΔE g) confirms a strong interaction between N2O and BeONTs, and an increase in conductivity of nanotube can propose that the 1Ca- and 2Ca-doped BeONTs are good candidates for making sensor for N2O gas. Upon N2O gas adsorption, the HOMO and LUMO levels moved to higher energies and the energy gap and other electrical properties of nanotube changed. Comparing results of dipole moment and the stabilization energy show that with increasing E (2) the dipole moment decrease and at the all adsorption models with doping 1Ca and 2Ca atoms the dipole moment increases significantly from original values.
References
Overview of greenhouse gases––nitrous oxide, US EPA, 164 (2014)
Iwamoto, M., Hamada, H.: Removal of nitrogen monoxide from exhaust gases through novel catalytic processes. Catal. Today 10, 57–71 (1991)
Kaptein, F., Rodriguez-Mirasol, J., Moulijn, J.A.: Heterogeneous catalytic decomposition of nitrous oxide. Appl. Catal. B Environ. 9, 25–64 (1996)
Kondratenko, E.V., Pérez-Ramirez, J.: Micro-kinetic analysis of direct N2O decomposition over steam-activated. Fe-silicalite from transient experiments in the TAP reactor. Catal. Today 121, 197–203 (2007)
U.S. Greenhouse gas inventory report: 1990–2013, US EPA. 60 (2015)
CFR Part 98––revisions to the greenhouse gas reporting rule and final confidentiality US EPA. Environmental Protection Agency. (2014)
Steinfeld, H., Gerber, P., Wassenaar, T., Castel, V., Rosales, M., de Haan, C.: Livestock’s long shadow––Environmental issues and options. (2006)
Nitrous oxide emissions. US Environmental Protection Agency (2016)
Sources of greenhouse gases. IPCC TAR WG1 2001 (2012)
Jeong, T.S., Yu, J.H., Mo, H.S., Kim, T.S., Youn, C.J., Hong, K.J.: Detection mechanism and characteristics of ZnO-based N2O sensors operating with photons. J. Korean Phys. Soc. 63, 2012–2017 (2013)
Kanazawa, E., Sakai, G., Shimanoe, K., Kanmura, Y., Teroaka, Y., Miura, N., Yamazoe, N.: Metal oxide semiconductor N2O sensor for medical use. Sens. Actuators B 77, 72–77 (2001)
Kantorovich, L.N., Gillan, M.J.: The energetics of N2O dissociation on CaO(001). Surf. Sci. 376, 169–176 (1997)
Karlsen, E.J., Nygren, M.A., Pettersson, L.G.M.: Theoretical study on the decomposition of N2O over rocksalt metal-oxides MgO-BaO. J. Phys. Chem. A 106, 7868–7875 (2002)
Xu, Y.J., Li, J.Q., Zhang, Y.F.: Conversion of N2O to N2 on MgO (001) surface with vacancy: a DFT study. Chin. J. Chem. 21, 1123–1135 (2003)
Lu, X., Xu, X., Wang, N.Q., Zhang, Q.E.: N2O decomposition on MgO and Li/MgO catalysts: a quantum chemical study. J. Phys. Chem. B 103, 3373–3379 (1999)
Karlsen, L.G., Pettersson, M.: N2O decomposition over BaO, including effects of coverage. J. Phys. Chem. B 106, 5719–5721 (2002)
Snis, A., Stromberg, D., Panas, I.: N2O adsorption and decomposition at a CaO(100) surface, studied by means of theory. Surf. Sci. 292, 317–324 (1993)
Ovideo, J., Sanz, J.F.: N2O decomposition on TiO2 (110) from dynamic first-principles calculations. J. Phys. Chem. B 109, 16223–16226 (2005)
Solans-Monfort, X., Sodupe, M., Branchadell, V.: Spin-forbidden N2O dissociation in Cu–ZSM-5. Chem. Phys. Lett. 368, 242–246 (2003)
Andelman, B.J., Beutel, T., Lei, G.D., Sachtler, W.M.H.: On the mechanism of selective NO x reduction with alkanes over Cu/ZSM-5 Appl. Catal. B Environ. 11, 1–9 (1996)
Chen, H.J., Matsuoka, M., Zhang, J.L., Anpo, M.: Investigations on the effect of Mn ions on the local structure and photocatalytic activity of Cu(I)-ZSM-5 catalysts. J. Phys. Chem. B 110, 4263–4269 (2006)
Zhanpeisov, N.U., Ju, W.S., Matsuoka, M., Anpo, M.: Quantum chemical calculations on the structure and adsorption properties of NO and N2O on Ag+ and Cu+ ion-exchanged zeolites. Struct. Chem. 14(3), 247–255 (2003)
Kachurovskaya, N.A., Zhidomirov, G.M., Hensen, E.J.M., Santen, R.A.v.: Cluster model DFT Study of the intermediates of benzene to phenol oxidation by N2O on FeZSM-5 zeolites. Catal. Lett. 86, 25–31 (2003)
Kaucky, D., Sobalik, Z., Schwarze, M., Vondrova, A., Wichterlova, B.: Effect of FeH-zeolite structure and Al-Lewis sites on N2O decomposition and NO/NO2-assisted reaction. J. Catal. 238(2), 293–300 (2006)
Heyden, A., Peters, B., Bell, A.T., Keil, F.J.: J Comprehensive DFT study of nitrous oxide decomposition over Fe-ZSM-5. Condens. Matter Mater. Surf. Interfaces Biophys. 109(10), 4801–4804 (2005)
Kokalj, A., Kobal, I., Matsushima, T.: A DFT study of the structures of N2O adsorbed on the Pd (110) surface. J. Phys. Chem. B 107(12), 2741–2747 (2003)
Delabie, A., Pierloot, K.: The reaction of Cu(I) (1S and 3D) with N2O: an ab initio study. J. Phys. Chem. A 106, 5679–5685 (2002)
Wang, G.J., Jin, X., Chen, M.H., Zhou, M.F.: Matrix isolation infrared spectroscopic and theoretical study of the copper (I) and silver (I)–nitrous oxide complexes. Chem. Phys. Lett. 420, 130–134 (2006)
Baei, M.T., Soltani, A.R., Varasteh Moradi, A., Tazikeh Lemeskic, E.: Adsorption properties of N2O on (6,0), (7,0), and (8,0) zigzag single-walled boron nitride nanotubes: a computational study. Comput. Theor. Chem. 970, 30–35 (2011)
Baei, M.T., Soltani, A.R., Varasteh Moradi, A., Moghimi, M.: Adsorption properties of N2O on (6,0), (7,0), (8,0), and Al-doped (6,0) zigzag single-walled carbon nanotubes: a density functional study. Monatshefte Chem. 142, 573–578 (2011)
Soltani, A., Ramezani Taghartapeh, M., Tazikeh Lemeski, E., Abroudi, M., Mighani, H.: A theoretical study of the adsorption behavior of N2O on single-walled AlN and AlP nanotubes. Superlattices Microstruct. 58, 178–190 (2013)
Stelmachowski, P., Zasada, F., Piskorz, W., Kotarba, A., Paul, J.F., Sojka, Z.: Experimental and DFT studies of N2O decomposition over bare and Co-doped magnesium oxide—insights into the role of active sites topology in dry and wet conditions. Catal. Today 137, 423–428 (2008)
Wanbayor, R., De, P., Frauenheim, T., Ruangpornvisuti, V.: First-principles investigation of adsorption of N2O on the anatase TiO2 (101) and the CO pre-adsorbed TiO2 surfaces. Comput. Mater. Sci. 58, 24–30 (2012)
Nayebzadeh, M., Soleymanabadi, H., Bagher, Z.: Adsorption and dissociation of nitrous oxide on pristine and defective BeO and ZnO nanotubes: DFT studies. Monatshefte Chem. 145, 1745–1752 (2014)
Continenza, A., Wentzcovitch, R.M., Freeman, A.J.: Theoretical investigation of graphitic BeO. Phys. Rev. B 41, 3540–3544 (1990)
Baumeier, B., Kruger, P., Pollmann, J.: Structural, elastic, and electronic properties of SiC, BN, and BeO nanotubes. Phys. Rev. B 76, 085407(1)–085407(10) (2007)
Wu, W., Lu, P., Zhang, Z., Guo, W.: Electronic and magnetic properties and structural stability of BeO sheet and nanoribbons. ACS Appl. Mater. Interfaces 3, 4787–4795 (2011)
Wang, Y.Y.: Ding electronic structure of fluorinated and hydrogenated beryllium monoxide nanostructures. Phys. Status Solidi Rapid Res. Lett. 6, 83–85 (2012)
Gorbunova, M.A., Shein, I.R., Makurin, Y.N., Ivanovskaya, V.V., Kijko, V.S., Ivanovskii, A.L.: Electronic structure and magnetism in BeO nanotubes induced by boron, carbon and nitrogen doping, and beryllium and oxygen vacancies inside tube. Phys. E 41, 164–168 (2008)
Seif, A., Zahedi, E.: A DFT studies of structural and quadrupole coupling constants properties in C-doped BeO nanotubes. Superlattices Microstruct. 50, 539–548 (2011)
Fathalian, A., Kanjouri, F., Jalilian, J.: BeO nanotube bundle as a gas sensor. Superlattices Microstruct. 60, 291–299 (2013)
Ahmadi Peyghan, A., Yourdkhani, S.: Capture of carbon dioxide by a nanosized tube of BeO: a DFT study. Struct. Chem. 25, 419–426 (2014)
Ahmadaghaei, N., Noei, M.: Density functional study on the sensing properties of nano-sized BeO tube toward H2S. J. Iran. Chem. Soc. 11, 725–731 (2014)
Schmidt, M.W., Baldridge, K.K., Boatz, J.A., Elbert, S.T., Gordon, M.S., Jensen, J.H., Koseki, S., Matsunaga, N., Nguyen, K.A., Su, S.J., Windus, T.L., Dupuis, M., Montgomery, J.A.: General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363 (1993)
Parr, R.G., Donnelly, R.A., Levy, M., Palke, W.E.: Electronegativity the density functional viewpoint. J. Chem. Phys. 68, 3801–3807 (1978)
Parr, R.G., Szentpaly, L., Liu, S.: Electrophilicity index. J. Am. Chem. Soc. 121, 1921–1924 (1999)
Koopmans, T.: Über die Zuordnung von Wellenfunktionen und Eigenwerten zuden Einzelnen Elektronen Eines Atoms. Physica 1, 104 (1933)
Rezaei-Sameti, M., Samadi Jamil, E.: The adsorption of CO molecule on pristine, As, B, BAs doped (4,4) armchair AlNNTs: a computational study. J. Nanostruct. Chem. 6, 197–205 (2016)
Rezaei Sameti, M., Kazmi, A.: A computational study on the interaction between O2 and pristine and Ge-doped aluminum phosphide nanotubes. Turk. J. Phys. 39, 128–136 (2015)
Rezaei-Sameti, M., Yaghoobi, S.: Theoretical study of adsorption of CO gas on pristine and AsGa-doped (4, 4) armchair models of BPNTs. Comput. Condens. Matter 3, 21–29 (2015)
Scrocco, E., Tomasi, J.: Electronic molecular structure, reactivity and intermolecular forces: an euristic interpretation by means of electrostatic molecular potentials. Adv. Quantum Chem. 103, 115–193 (1978)
Luque, F.J., Lopez, J.M., Orozco, M.: Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 103, 343–345 (2000)
Scrocco, E., Tomasi, J.: The electrostatic molecular potential as a tool for the interpretation of molecular properties, the series Topics in Current Chemistry Fortschritte der Chemischen. Forschung 42, 95–170 (2005)
Li, Y., Liu, Y., Wang, H., Xiong, X., Wei, P., Li, F.: Synthesis, crystal structure, vibration spectral, and DFT studies of 4-aminoantipyrine and its derivatives. Molecules 18, 877–893 (2013)
Foster, J.P., Weinhold, F.: Natural hybrid orbitals. J. Am. Chem. Soc. 102, 7211–7218 (1980)
James, C., Amal Raj, A., Raghunathan, R., Hubert Joe, I., Jayakumar, V.S.: Structural conformation and vibrational spectroscopic studies of 2,6-bis(p-N, N-dimethyl benzylidene) cyclohexanone using density functional theory”. J. Raman Spectrosc. 12, 1381–1392 (2006)
Politzer, P., Truhlar, D.G.: Chemical applications of atoms and molecular electrostatic potential. Plessum Press, New York (1981)
Subash chandrabose, S., Krishnana, A.R., Saleem, H., Paramashwari, R., Sundaraganesan, N., Thanikachalam, V., Maikandan, G.: Vibrational spectroscopic study and NBO analysis on bis (4-amino-5-mercapto-1, 2, 4-triazol-3-yl) methane using DFT method. Spectrochim. Acta 77A, 877–884 (2010)
Arjunan, V., Raj, A., Ravindran, P., Mohan, S.: Structure–activity relations of 2-(methylthio) benzimidazole by FTIR, FT-Raman, NMR, DFT and conceptual DFT methods. Mol. Biomol. Spectrosc. 118, 951–965 (2014)
Liu, J.N., Chen, Z.R., Yuan, S.F., Zhejiang, J.: Study on the prediction of visible absorption maxima of azobenzene compounds. J. Zhejiang Univ. Sci. B 6, 584–589 (2005)
Acknowledgments
The author thanks the computational information center of Malayer University for providing the necessary facilities to carry out the research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rezaei-Sameti, M., Hemmati, N. N2O interaction with the pristine and 1Ca- and 2Ca-doped beryllium oxide nanotube: a computational study. J Nanostruct Chem 6, 343–355 (2016). https://doi.org/10.1007/s40097-016-0206-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-016-0206-1