
Abstract
The hopcalite (CuMnOx) catalyst is a well-known catalyst for CO oxidation at a low temperature and it is synthesized by the co-precipitation method with different types of precursors. Activity of the CuMnOx catalysts for CO oxidation is strongly dependent upon the combination of precursors, ranking in order {Mn(Ac)2 + Cu(NO3)2} > {Mn(Ac)2 + Cu(Ac)2} > {Mn(NO3)2 + Cu(NO3)2} > {Mn(NO3)2 + Cu(AC)2}. All the precursors were precipitated by KMnO4 solution and the precursors mostly comprised of MnO2, Mn2O3 and CuO phases. Keeping the same precipitant while changing the precursors caused a change in the lattice oxygen mobility which influenced the CO oxidation activity. The calcination strategy of the precursors has great influence on the activity of resulting catalysts. The reactive calcination (RC) conditions produce multifarious phenomena of CO oxidation and the precursor decomposition in a single-step process. The activity order of the catalysts for CO oxidation was as follows: reactive calcination (RC) > flowing air > stagnant air. Therefore, we recommended that the RC route was the more appropriate calcination route for the production of highly active CuMnOx catalysts. All the catalysts were characterized by X-ray diffraction, Fourier transform infrared spectroscopy, Brunauer–Emmett–Teller analysis, X-ray photoelectron spectroscopy and scanning electron microscopy technique. The influence of precursors on the structural properties and the catalytic activity of co-precipitation derived binary CuMnOx catalysts for CO oxidation has been investigated.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Carbon monoxide (CO) is a poisonous gas that needs to be removed from the air. It is a colorless, odorless, tasteless and non-irritating gas, which makes it very difficult for humans to detect. Huge amounts of CO are emitted in the world, mainly from transportation, power plants, industrial and domestic activities [1, 2]. The transportation sector contributes the largest source of CO emissions in the developed countries. In comparison to a diesel engine, the petrol engine vehicle produces more CO into the environment [3, 4]. CO is formed in an internal combustion (I.C.) engine that is operated by the burning of fossil fuels (petrol or diesel), as an intermediate reaction during the incomplete combustion of HC [5]. The low temperature catalytic oxidation of CO is a very important process and it is essential in many applications such as in residential and automotive air-cleaning technologies, CO detectors, gas masks for firefighters, mining application and selective oxidation of CO in reformer gas for the fuel cell applications [6, 7]. The performance of catalytic converter is highly dependent upon the type of catalyst was used. In the presence of a catalyst, the rate of chemical reaction is increase; it acts like an agent that reduces the activation energy of the reactions [8]. The noble metals (Pt, Pd, Rh, Au, etc.) and base metals (Cu, Mn, Cr, Co, Ni, Fe, etc.) were widely used as catalysts in the catalytic converter [9, 10]. Commercial catalysts that were mainly applied for CO oxidation present in the exhaust gas were noble metals. It has a high activity and thermal stability, so it was widely used as a catalyst in catalytic converter [11]. However, the employment of noble metal catalysts was limited due to their low availability, sulfur poisoning, and high cost [12]. Therefore, it was important to find an alternative catalytic component to replace the noble metal catalyst and attention has been given to base metals as catalysts.
Compared to noble metal catalysts, the hopcalite (CuMnOx) is one of the oldest known catalysts for CO oxidation at a low temperature. It is widely used for the respiratory protection systems in many types of applications like military, mining, and space devices, etc. The other practical advantages of hopcalite catalysts are cost efficiency, high availability, moisture resistance and high catalytic activity that results from their ability to provide an active oxygen species by changing oxidation states [13, 14]. In 1920 Lamb, Bray and Frazer discovered that various mixed oxides of Cu, Mn, Ag, and Co, and identified them as a group of catalysts known as hopcalite (CuMnOx) [15]. The structure of hopcalite catalyst is strongly dependent upon the preparation methods, (Cu:Mn) molar ratio, drying temperature and calcination conditions of the catalyst [16]. When oxygen species associated with copper in the CuMnOx catalyst was very active for the low temperature catalytic oxidation of CO [17]. The lattice oxygen mobility, specific surface area, and pore volume were highly effective on the activity of the catalysts for CO oxidation. To increase the reactivity of lattice oxygen associated with copper species as well as the mobility of lattice oxygen from the manganese species [18]. The CuMnOx catalyst has a high catalytic activity in CO oxidation that could be ascribed to the resonance system \({\text{Cu}}^{ 2+ } \, + \,{\text{Mn}}^{ 3+ } \rightleftarrows {\text{Cu}}^{ + } \, + \,{\text{Mn}}^{ 4+ }\) and the high adsorption of CO onto Cu2+/Mn4+ and of O2 onto Cu+/Mn3+ [19]. The Cu oxide was found to be poorly active for CO oxidation, but in conjunction with Mn oxide in appropriate proportions, some very active catalyst systems were generated [20].
Till date, there are various methods (i.e., co-precipitation, supercritical antisolvent precipitation, sol–gel, ultrasonic aerosol pyrolysis, and reduction) that have been implemented to prepare the CuMnOx catalysts. Among these methods, the conventional co-precipitation method can be use to produce the CuMnOx catalysts with high activity [21]. The screening of best precursor and successive calcination strategies of CuMnOx catalysts enhances their activity for CO oxidation [22]. Thus, the state of precursor varies depending on the types of chemicals that are use, which results in the development of calcined catalysts with different crystalline phases and different catalytic performances [23]. The high activity is due to the formation of Cu–Mn spinel CuMn2O4 formed during the co-precipitation process. The redox reaction has been proposed to explain that the CuMnOx catalyst activity, i.e., an electronic transfer between copper and manganese cations within the spinel lattice [24]. The occurrence of individual phases and their quantitative proportions depend upon the temperature, pH and concentrations of the reactant solutions [25].
According to Hutching et al., the performance of CuMnOx catalysts very much depends upon the calcination conditions of the precursors and the subsequent pretreatment of the catalysts [26]. The high-temperature calcination causes sintering of the active crystallites with a consequent loss of surface area and subsequently adversely affects on the performance of the catalyst [27]. Therefore, to minimize the above-mentioned drawbacks of two steps of calcination and pretreatment, a newer route of single-step thermal treatment of the precursors in a reactive 4.5% CO–air mixture at a low temperature (160 °C) has been recently suggested by the authors for preparing highly active catalysts by passing the separate pretreatment step [28]. This single-step thermal treatment of the precursor is called “RC method”. It is postulated that the RC method parallel to multifarious phenomena of CO oxidation and precursor decomposition, causes a synergistic effect in the formation of oxygen-deficient catalyst at a low temperature. The success of CuMnOx catalyst has prompted a great deal of fundamental work devoted to clarifying the role played by each element and the nature of active sites. The activity of CuMnOx catalyst is highly dependent upon the structure and the preparation route [29]. Recently, Tang et al. have synthesized the nano-crystalline CuMnOx catalysts using the supercritical antisolvent precipitation method and found them to be more than twice as active as the conventionally prepared hopcalite catalysts for the CO oxidation [30]. They attributed the high catalytic activity to the nano-crystalline and homogeneous nature of the synthesized CuMnOx [31, 32]. The presence of manganese oxide is likely to facilitate the reduction of copper oxide, involving coordination between CuO and MnO2, the manganese oxide acting as an oxygen donor and copper oxide acting as the oxygen acceptor. A previous work demonstrated that the presence of Cu2+ and Mn3+ is essential for the high catalytic activity of CuMnOx catalyst [33, 34]. In present study, the different types of precursors have been used to change the crystalline phase of the catalysts and to find out the best CuMnOx catalyst, which will give the highest activity for CO oxidation.
Experimental
Catalyst preparation
The CuMnOx catalysts were prepared by the co-precipitation method. All the materials used for the preparation work were of analytical reagent (A.R.) grade. A solution of Mn(Ac)2 or Mn(NO3)2 (0.85 M) was added to 0.15 M of Cu(Ac)2 or Cu(NO3)2 and stirred for 1 h. The mixed solution was taken in the burette and added dropwise to a solution of KMnO4 (0.25 M) under the vigorous (continuous) stirring conditions for the co-precipitation purpose. The resultant precipitate was stirred continuously for 2 h. In the previous research, the optimum molar ratio of Cu:Mn in the CuMnOx catalyst was 1:8 [29]. The precipitates were filtered and washed several times with hot distilled water to remove all the anions. The cake thus obtained was dried at the temperature 110 °C for 24 h in the oven and calcined at 300 °C for 2 h. All the precursors were calcined in three different ways; first following the traditional method of calcination in stagnant air at 300 °C just above the decomposition temperatures of the precursors for 2 h in a muffle furnace, second in situ calcination in flowing air at a rate of 32.5 ml min−1 at 300 °C for 2 h. The third way calcination was carried out using the in situ reactive calcination (RC) as described. The reactive calcination of the precursors was carrying out in situ in a down-flow bench-scale tubular reactor with a defined amount of precursor that was diluted with α-alumina to make a total volume of 1 ml at atmospheric pressure. The nomenclature of the resulting catalysts thus formed was given by the first capital letter of the corresponding precursors use and the suffixes ‘SA’, ‘FA’ and ‘RC’ denote whether they were obtained by calcination in air, flowing air or by RC, respectively, as presented in Table 1.
Reactive calcination of the precursor was carried out by the introduction of a low concentration of chemically reactive CO–air mixture (4.5% CO) at a total flow rate of 32.5 ml min−1 over the hot precursors. The temperature of the reactor bed was increased from room temperature to 160 °C where CO conversion has started. This temperature was maintained for a defined period of time where the CO concentration was measured in the exit stream of the reactor at regular intervals until 100% CO conversion was achieved. After achieving total CO conversion, the resultant catalyst was annealed for half an hour at the same temperature, than the temperature was increased up to 300 °C and held for an hour followed by cooling down to room temperature in the same environment. The catalysts prepared as given above were stored in capped glass sample holders placed in desiccators.
Characterization of catalysts
The scanning electron micrograph (SEM) produced the high-resolution image of a catalyst by an electron beam, and the image of the catalyst is recorded on Zeiss EVO 18 (SEM) instrument. The magnification of images is 5000× and an accelerating voltage of 15 kV was applied. It provides information about the average aggregate size, crystalline degree and the microstructures of the catalyst. The energy dispersive X-ray analyzer (EDX) provides information about elemental identification and performs a quantitative composition analysis of compounds present in the catalyst. The X-ray diffraction (XRD) analysis of the catalyst is carried out by using Rigaku D/MAX-2400 diffractometer with Cu-Kα radiation at 40 kV and 40 mA. It is a fast analytical technique mainly applied for the measurement of phase identification, crystal orientation, crystallite size, unit cell dimensions and crystal defects, etc. The Fourier transform infrared spectroscopy (FTIR) provides information about the kind of materials present in a catalyst sample with their peak values. The measurement is done by the Shimadzu 8400 FTIR spectrometer in the range of 400–4000 cm−1. The Brunauer–Emmett–Teller analysis (BET) provides information about the specific surface area, pore volume and pore size of the catalyst. The isotherm is recorded by Micromeritics ASAP 2020 analyzer with the physical adsorption of N2 at the temperature of liquid nitrogen (− 196 °C) with an average pressure range of 0.05–0.30 P/Po.
Experimental procedure
The catalytic oxidation of CO over all the CuMnOx catalysts were carried out at atmospheric pressure in a fixed bed down-flow tubular reactor containing 100 mg of catalyst which was diluted to 1 ml of Al2O3 at a different temperature. The reactor was placed vertically in a split-open furnace in which the following reaction conditions were maintained: Catalyst particle size, 40–100 mesh, using a lean mixture of 2.5% CO (v/v) in the air, maintaining the total flow rate at 60 ml min−1. The details of the experimental apparatus setup are given in Fig. 1.

Experimental apparatus setup for testing the catalyst
The flow rate of gasses was monitored with the help of digital gas flow meter. The air feed was made free of moisture and CO2 by passing it through CaO and KOH pellet drying towers. The catalytic experiments were carried out under steady-state conditions. Typically, the reactor was heated to the desired temperature with the help of a microprocessor-based temperature controller. A temperature control of ± 0.5 °C was achieved. The gaseous products were analyzed by an online gas chromatograph (Nucon Series 5765) equipped with a methaniser, Porapack Q-column and FID detector for the concentration of CO and CO2. The catalytic activity was measured for the conversion of CO into CO2. The conversion of CO at any instant was calculated on the basis of values of the concentration of CO (CCO)in in the feed and the concentration of CO2 (CCO)out in the product stream. The concentration of CO was proportional to the area of chromatogram ACO (ACO)in that at any instant was proportional to the area of the chromatogram of CO2 (ACO)out formed.
Blank experiment
A blank experiment was carried out with Al2O3 only in place of catalyst. The bed temperature increased up to 300 °C, but practically no oxidation of CO has observed under the experimental conditions. The blank test shows that the performance of reactor in the absence of catalyst for CO oxidation with the increase of temperature does not show any activity for CO oxidation. Thus, the catalytic effect of the reactor wall and the alumina use as diluents can be neglected within the experimental conditions.
Catalyst characterization
Characterization of all the CuMnOx catalysts prepared by various precursors in reactive calcination conditions was done by the following techniques and activity of the catalyst for CO oxidation was discussed below.
Morphology analysis
The scanning electron micrograph (SEM) instrument was used for the microstructure analysis of the optimized CuMnOx (Cu1Mn8) catalysts. The SEM micrograph (Fig. 2) clearly shows the large differences in the microstructure and morphology of the CuMnOx catalysts formed by the different precursors. They all show granular particles between 0.30 and 10 µm calculated by “Image J software” with varying degrees of agglomeration as mentioned in Table 2. The digital image processing (DIP) has only been applied to the SEM analysis of coarse aggregates and fine aggregates. The image analysis instrument may report distributions based on particle length as opposed to spherical equivalency, and they may build volume distributions based on shapes other than spheres. SEM technique (magnification ranging from 20× to approximately 30,000×, spatial resolution of 50 to 100 nm). The SEM has more than 300 times the depth of field of the light microscope. Magnification will increase if we reduce the size of the area scanned on the specimen. The SEM images of particles may be characterized by having higher intensity than the surrounding pixels. The particles can also be characterized by their form, such as roundness, elongation and orientation. The image J software provides the histogram for particle size distribution. The histogram of bulk samples shows that the maximum size distribution is in the range of 0.315–1.130 µm [35, 36]. As shown in the SEM micrograph, the particles comprised grains of coarser, coarse, fine and finest sizes using CuMnRC4, CuMnRC3, CuMnRC2 and CuMnRC1 as catalysts, respectively. The SEM result was also in good agreement with XRD analysis. The average particle size of CuMnRC1, CuMnRC2, CuMnRC3 and CuMnRC4 catalyst was 0.315 µm, 0.516 µm, 0.891 µm and 1.125 µm, respectively.

SEM image of a CuMnRC4, b CuMnRC3, c CuMnRC2 and d CuMnRC1
The particles of CuMnRC1 catalyst was least agglomerated, highly porous, uniformly distributed, and had a higher surface area. The choice of precursors in the CuMnOx catalysts lead to the change of surface distribution of Cu, Mn and O elements, which might be related to the CO oxidation [36,37,38]. Thus, the different precursors used in the preparation of CuMnOx (Cu1Mn8) catalysts in the present study considerably affect the particle size as well as the morphology of resulting catalysts.
The synergetic effect of CuMnRC1 catalyst was highly dependent upon the catalyst composition and the nature of oxidized compounds. All the catalysts have different morphologies, indicating that the types of precursors used in the preparation of catalysts have a great influence on the catalytic activity. The mixing of {Mn(Ac)2 + Cu(NO3)2} and precipitation by KMnO4 solution was more efficient in improving the catalytic behavior of CuMnOx catalysts for CO oxidation.
Elemental analysis
To verify the atomic ratio of Cu to Mn in the CuMnOx catalysts, scanning electron microscopy (SEM) with energy dispersive X-Ray analysis (SEM–EDX) techniques were performed in a large scanning range randomly for the samples CuMnRC4, CuMnRC3, CuMnRC2 and CuMnRC1, respectively. The elemental concentration distribution of the catalyst granules was determined using Isis 300 software [39]. The result of SEM–EDX analysis has shown that all the catalyst samples were pure due to the presence of their relevant element peaks only. It was provided in an EDX mapping of Cu and Mn instead on different cross-sectioned marks of CuMnOx catalysts granules to determine the concentration of different elements located at different cross-sections on the catalysts’ granular surfaces as shown in the Fig. 3. Table 3 represents the relative atomic abundance of Cu, Mn and O species present in the surface layers of CuMnOx catalysts. The presence of Cu, Mn and O peaks on the surface of CuMnOx catalysts can be clearly detected.

SEM–EDX image of a CuMnRC4, b CuMnRC3, c CuMnRC2 and d CuMnRC1
The surface atomic ratio of the catalysts prepared with different precursors was compiled in Table 3. The atomic ratio and weight ratio of Cu/Mn in the CuMnRC1 catalyst was approximately 0.155 and 0.171, respectively. The molar ratio of Cu/Mn in all the catalyst samples using co-precipitation method was approximately 0.187 and it was very close to the actual dosage of Cu and Mn percentage present in the precursors.
The abundant surface oxygen atoms of the CuMnRC catalyst can react with the absorbed CO and thus lead to better catalytic activity in the Mars–van Krevelen type mechanism (MvK) which was frequently suggested for the metal oxides [40]. It was clear from the Table 3 and Fig. 3 that the atomic percentage and weight percentage of Mn was also higher in comparison to Cu and O. A calcination strategy of CuMnRC catalyst has highly influenced the elemental distribution of different elements present on the catalysts’ surfaces. The choice of precursors can lead to the change of surface distribution of Cu, Mn and O elements, which might be related to the CO oxidation on the CuMnRC catalysts. The presence of a small amount of oxygen in the surface layer of CuMnRC1 catalyst causes the number of active sites present on the catalyst surface to increase. The successful redox reactions in the presence of lattice oxygen of CuMnOx catalysts and its charge (hole or electron) transfer ability between Cu+ and Cu2+ [39]. The surface property of the catalyst was highly influenced by the calcination process; the crystallinity was lost with the increase of calcination temperature, with gradual transformation to amorphous CuMnOx phase.
Phase identification and cell dimensions
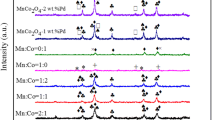
The phase identification and cell dimensions of CuMnOx catalysts prepared in reactive calcination conditions were done by the X-ray powder diffraction (XRD) technique. The crystalline size and coordinate dimensions present on the surface layer of the catalysts were identified. The mean crystallite size (d) of the catalyst was calculated from the line broadening of the most intense reflection using the Scherrer Equation (d = 0.89λ/βcosθ). Where d was the mean crystallite diameter, 0.89 was the Scherrer constant, l is the X-ray wave length (1.54056 Å), and b was the effective line width of the observed X-ray reflection. It provides information about the structure, phase, crystal orientation, lattice parameters, crystallite size, strain and crystal defects, etc., [32]. XRD patterns of the CuMnOx catalyst produced by reactive calcination of the various precursors (CuMnRC1, CuMnRC2, CuMnRC3 and CuMnRC4) are displayed in Fig. 4. XRD pattern of the CuMnRC1 catalyst has shown that the diffraction peak at 2θ of 32.96 corresponds to its lattice plane (112) end-centered cubic Cu1Mn8O2 (PDF-75-1010 JCPDS file). The crystallite size of the catalyst was 3.90 nm. In reactive calcination of CuMnRC2 catalyst was shown that the diffraction peak at 2θ of 33.60 corresponds to its lattice plane (110) of face-centered cubic Cu0.2Mn1.8O4 (PDF-72-1254 JCPDS file). The crystallite size of the catalyst was 4.215 nm.

XRD analysis of a CuMnRC1, b CuMnRC2, c CuMnRC3 and d CuMnRC4
The XRD pattern of the CuMnRC3 catalyst has shown that the diffraction peak at 2θ of 32.43 corresponds to its lattice plane (111) of cubic-centered Cu1Mn6.8O4 (PDF-84-0543 JCPDS file). The crystallite size of the catalyst was 5.61 nm. XRD pattern of the CuMnRC4 catalyst has shown that the diffraction peak at 2θ of 33.23 corresponds to its lattice plane (121) body-centered tetragonal Cu0.2Mn1.8O2 (PDF-78-1012 JCPDS file). The crystallite size of the catalyst was 7.90 nm. The broader peak in CuMnRC1 has indicated that the relatively amorphous nature of the catalyst and their structure, phase, and crystallite size are also discussed in Table 4.
After XRD analysis, we have confirmed that the crystallite size of CuMnRC1 is lower than the CuMnRC2, CuMnRC3 and CuMnRC4 catalysts. The structure, phase and crystallite size of CuMnRC catalysts have been represented in Table 4. The crystallite size of the particles present in a catalyst surface obtained by RC conditions was as follows: CuMnRC4 > CuMnRC3 > CuMnRC2 > CuMnRC1. It was quite apparent that the crystallite size of CuMnRC1 catalyst exhibited the smallest size (3.90 nm) in comparison to CuMnRC2 (4.21 nm), CuMnRC3 (5.61 nm) and CuMnRC4 (7.90 nm). From Table 4 and Fig. 4, we have confirmed that the particles present in CuMnRC1 catalyst were mostly in crystalline form, and producing narrow size high-intensity diffraction lines, as compared to the other catalysts. The refinement of the XRD pattern of CuMnRC sample has shown that there will be no impurity phases present in the catalysts.
Identification of the materials present in a catalyst
The identification of the metal–oxygen bonds present in the catalyst was done by the Fourier transform infrared spectroscopy (FTIR) analysis. The different peaks show various types of chemical groups present on the catalyst surfaces. The FTIR transmission spectrum of the CuMnRC1, CuMnRC2, CuMnRC3 and CuMnRC4 catalysts prepared by reactive calcination (RC) conditions is shown in the Fig. 5. In the CuMnRC1 catalyst at the transmittance conditions, there were total three peaks we obtained, the IR band (1640 cm−1) has shown the presence of MnO2 group, (3490 cm−1) CuO group and (1180 cm−1) H–OH group. In the CuMnRC2 catalyst at the transmittance conditions, there were total five peaks we obtained, the MnO2 vibration mode was observed at (1640 cm−1), (3490 cm−1) was assigned to CuO group, (2350 cm−1 and 1180 cm−1) H–OH group and (2120 cm−1) C=O group.

FT-IR analysis of a CuMnRC1, b CuMnRC2, c CuMnRC3 and d CuMnRC4
In the CuMnRC3 catalyst at the transmittance conditions, there were a total of four peaks that we obtained, the IR band (1640 cm−1) has shown the presence of MnO2 group, (3490 cm−1) CuO group, (1180 cm−1) H–OH group and (2140 cm−1) CO32− group. In the CuMnRC4 catalyst at the transmittance conditions, there were a total of four peaks that we obtained, the MnO2 vibration mode was observed at (1640 cm−1) due to the stretching of Mn–O bond and transmission spectra at (3530 cm−1) was assigned to CuO group. The other phases like CO32− and C=O were present at (1330 cm−1) and (2920 cm−1), respectively. The spectra of impurities decrease in the following order: CuMnRC4 > CuMnRC3 > CuMnRC2 > CuMnRC1. The best result we can get from the FTIR analysis was that the CuMnRC1 catalyst has the highest purity as compared to the other catalysts; therefore, we get the best activity results for CO oxidation.
Identification and quantification of elements
The surface valence state and chemical concentration of the catalysts were investigated by the X-ray photoelectron spectroscopy (XPS) analysis. The XPS analysis that was mainly used to understand the physical and chemical changes of the catalyst with exposure to gaseous molecules under the different thermal conditions has been examined. Although, it can be proposed that the high binding energy was preferable for the CO oxidation. The XPS spectra in the Cu(2p), Mn(2p) and O(1s) regions are shown in the figures. The prominent peak of Cu(2p) level in CuMnRC1, CuMnRC2, CuMnRC3 and CuMnRC4 catalyst was deconvoluted into five to six peaks centered as shown in the Fig. 6. By performing peak fitting deconvolution, the main Cu(2p) in all the catalyst samples was found to be Cu(II) oxide form. The binding energy of Cu(2p) in CuMnRC1 catalyst was 954.033 eV, 956.600 eV, 960.844 eV, 962.22 eV, 964.718 eV and 967.32 eV, respectively, and the highest binding energy peak of Cu(2p) in CuMnRC1 catalyst was obtained at 964.718 eV. The binding energy of Cu(2p) in CuMnRC2 catalyst was 954.019 eV, 956.512 eV, 960.714 eV, 962.102 eV, 964.523 and 967.149 eV, respectively, and the highest binding energy peak of Cu(2p) in CuMnRC2 catalyst was obtained at 964.523 eV.

XPS analysis of Cu(2p) in a CuMnRC1, b CuMnRC2, c CuMnRC3 and d CuMnRC4
The binding energy of Cu(2p) in CuMnRC3 catalyst was 954.19 eV, 956.24 eV, 960.116 eV, 964.291 eV and 967.176 eV, respectively, and the highest binding energy peak of Cu(2p) in CuMnRC3 catalyst was obtained at 964.291 eV. The binding energy of Cu(2p) in CuMnRC4 catalyst was 954.02 eV, 956.17 eV, 960.116 eV, 964.158 eV and 967.131 eV, respectively, and the highest binding energy peak of Cu(2p) in CuMnRC4 catalyst was obtained at 964.158 eV. The highest binding energy peak of Cu(2p) in CuMnRC1, CuMnRC2, CuMnRC3 and CuMnRC4 catalyst was 964.718 eV, 964.523 eV, 964.291 eV and 964.158 eV, respectively. It was clear from the Table 5 and Fig. 7 that the binding energy of Cu(2p) in CuMnRC1 catalyst was highest as comparison to other catalysts. The prominent peak of Mn(2p) level in CuMnOx catalyst was deconvoluted into double peak and it is represented in the Fig. 7.

XPS analysis of Mn(2p) in a CuMnRC1, b CuMnRC2, c CuMnRC3, d CuMnRC4
By performing peak fitting deconvolution of the main Mn(2p) in all the catalyst samples, three main components including Mn4+, Mn3+ and satellite can be found. The difference between the binding energy values of Mn4+ and Mn3+ ions was small. The binding energy of Mn(2p) in CuMnRC1, CuMnRC2, CuMnRC3 and CuMnRC4 catalyst at reactive calcination condition was (643.212 eV and 655.361 eV), (643.189 eV and 655.349 eV), (643.172 and 655.295 eV) and (643.151 eV and 655.285 eV), respectively, and it will be associated with the presence of Mn3+, Mn4+ and satellite in all the samples. The highest binding energy peak of Mn(2p) in CuMnRC1, CuMnRC2, CuMnRC3 and CuMnRC4 catalyst was 655.361 eV, 655.349 eV, 655.295 eV and 655.285 eV, respectively. The broad Mn4+ peak present in CuMnRC1 catalyst was higher than CuMnRC2, CuMnRC3 and CuMnRC4 catalyst. Finally, we confirmed that the binding energy of Mn(2p) present in CuMnRC1 catalyst was highest as compared to other catalyst samples.
In present study, the oxygen with the binding energy of 529.58 eV was the main form and could be assigned to the chemisorbed oxygen (Oa). The presence of lattice oxygen was very small in CuMnRC1 catalyst. The binding energy of O(1s) in CuMnRC1, CuMnRC2, CuMnRC3 and CuMnRC4 catalyst was 529.84 eV, 529.41 eV, 529.24 eV and 529.08 eV, respectively. The chemical state of Cu, Mn and O in CuMnRC catalyst was Cu(II) Oxide, Mn2O3 and C–O form and it was represented in Table 5. One of noticeable facts was that the amount of oxygen is present less in reactive calcined prepared CuMnRC1 catalyst as compared to the CuMnRC2, CuMnRC3 and CuMnRC4 catalysts, due to an absence of lattice oxygen which creates oxygen vacancies for CO oxidation. The content order of Oa/(Oa + Ol) ratio was shown as follows: CuMnRC1 > CuMnRC2 > CuMnRC3 > CuMnRC4. The presence of higher oxidation state phases could be the result of a greater degree of surface interaction between the easily oxidisable manganese phase and highly reducible copper phase [41]. The high amount of surface chemisorbed oxygen (most active oxygen) was more preferable for enhancing the catalytic activity for CO oxidation.
Surface area measurement of catalyst
BET surface area of CuMnOx catalyst was prepared by the co-precipitation method with a novel route of reactive calcination was much superior to the other calcination routes like flowing air and stagnant air. Clearly, the textural property of CuMnRC1 catalyst was much superior to the CuMnRC2, CuMnRC3 and CuMnRC4 catalysts. It can be visualized from the Table 6 and Fig. 8 that the pore volume and pore size of CuMnRC1 catalyst in reactive calcined was much higher than the other catalyst samples. The isotherm gave useful information on the mesopores’ structure through its hysteresis loop. The catalyst samples exhibited hysteresis loop, which indicated that the pores were exhibiting geometries of mesopores. The surface area and pore volume isotherms of CuMnRC catalyst are shown in the Fig. 8. The existence of hysteresis loop at a relative pressure (P/P0) of 0.8–1.0 indicates the porosity arising from the non-crystalline intra-aggregate voids and spaces formed by inter-particle contacts.

Textural properties a N2 adsorption–desorption isotherms and b Pore size distributions
The N2 adsorption–desorption isotherms and their porous textural parameters, such as specific surface area (SBET) and pore volume (Vtotal) are also listed in Table 6. The surface area of CuMnRC1, CuMnRC2, CuMnRC3 and CuMnRC4 catalyst was 118.45, 107.81, 95.28 and 88.57 m2/g, respectively. These data clearly indicate that the precursors have also a high efficacy for the low temperature CO oxidation. In the mesopores, molecules from a liquid-like adsorbed phase having a meniscus of which curvature was associated with the Kelvin equation, providing the pore size distribution calculation. Specific surface area and total pore volume were two major factors which can influence the catalytic activity for CO oxidation.
The catalyst surface areas of similar magnitude regardless of the preparation atmosphere; however, there was a general increase in surface area as a result of choosing suitable precursors.
The average pore diameter was increased with the increase of calcination temperature because a high-temperature treatment led to particle sintering accompanied by a loss in the active area. The CuMnRC1 catalyst surface area (118.45 m2/g) and pore volume (0.627 cm3/g) were very high, so that it was most active for CO oxidation at a low temperature. A large number of more pores present in a catalyst surface means a higher number of CO molecules captured on their surfaces, therefore, it has to shows the better catalytic activity. The specific surface area was measure by BET analysis and it was also followed by the SEM and XRD results.
Catalyst performance and activity measurement
Activity test of the catalyst was carried out to evaluate the effectiveness of CuMnOx catalysts as a function of temperature. It was evaluated in different calcination conditions like stagnant air, flowing air and reactive calcination. The activity was increased with the increase of temperature from room temperature to certain high temperature for full conversion of CO. Activity of the CuMnOx catalysts, produced by co-precipitation method using the different precursors, for the CO oxidation was discussed below. All the catalysts were highly active for CO oxidation. The improved catalytic activity of the catalysts can be ascribed to the unique structural, textural characteristics and the smallest crystallite size. The light-off characteristics were used to evaluate the activity of resulting catalysts with the increase of temperature. The characteristic temperature T10, T50 and T100 correspond to the initiation of oxidation, half conversion and full conversion of CO, respectively.
Stagnant air calcination
An activity of the CuMnSA catalysts was measured in the laboratory at stagnant air calcination conditions for CO oxidation. The CO oxidation by CuMnSA catalyst was highly influenced by a combination of factors including preparation conditions, drying temperature, calcination conditions and the presence of Cu2+, Mn2+ and O on the surface of catalyst. The stagnant air calcination of CuMnOx precursor was done in a muffle furnace in presence of stagnant air. A clean surface was exposed by a mixture of CO and air quickly becomes covered with CO since it requires single vacant adsorption sites. The catalytic activity tests were carried out to evaluate the activity of resulting catalysts as a function of temperature. The interaction between various chemical compounds (Cu–Nitrate or Cu–Acetate and Mn–Nitrate or Mn–Acetate) with the precipitation agent KMnO4 as the formation of a highly disordered mixed metal oxide was the cause of high catalytic activity for CO oxidation.
It was evident from the Table 7 and Fig. 9 that the oxidation of CO was just initiated near around the room temperature at 25 °C and 50% conversion of CO has occurred at 90 °C over CuMnSA1 catalyst, which was less by 10 °C, 20 °C and 30 °C over than that of CuMnSA2, CuMnSA3 and CuMnSA4 catalyst, respectively. The total oxidation temperature of CO was 180 °C for CuMnSA1, which was less by 20 °C, 30 °C and 50 °C over than that of CuMnSA2, CuMnSA3 and CuMnSA4 catalysts, respectively. Finally, we confirmed that the CuMnSA1 catalyst has shown that the best catalytic activity towards CO oxidation as compared to the other catalysts.

Catalytic activity of CuMnSA catalysts for CO oxidation
The activity order of each catalyst for CO oxidation was in accordance with their characterization. The order of activity of different types of CuMnOx catalysts for CO oxidation was as follows: CuMnSA1 > CuMnSA2 > CuMnSA3 > CuMnSA4. Using the same (KMnO4) precipitant, the catalysts prepared with a combination of ({Mn(Ac)2 + Cu(NO3)2} as the precursors were considerably more effective than the other catalysts. An activity of the multiphase catalysts CuO and MnO2 were several times higher than those of single-phase oxides CuMnOx catalysts, especially in the lower temperature range. The coordination between Mn oxide and Cu oxide in an appropriate proportion with the CuMnOx catalyst improved their performance for CO oxidation.
Flowing air calcination
The copper oxide and manganese oxide were most widely used as precursors in the preparation of various catalysts. The individual property of Cu oxide and Mn oxide in CuMnOx has also an effect on the activity of resulting catalysts [42]. In the flowing air calcination conditions, a fresh catalyst was used to measure the activity of resulting catalysts at each temperature. The increase in local temperature will decrease the decomposition of the precursor. In the initial conditions, a very slow exothermic reaction for CO oxidation was going on over the catalyst, which causes a rise in the local temperature. The oxidation of CO was just initiated at 25 °C in flowing air calcination conditions over all the catalyst samples and 50% conversion of CO over the CuMnFA1 catalyst was 70 °C, which was less by 10 °C, 30 °C and 50 °C over than that of CuMnFA2, CuMnFA3 and CuMnFA4 catalysts, respectively.
The total oxidation temperature of CO was 120 °C for CuMnFA1 catalyst, which was less by 30 °C, 55 °C and 70 °C over than that of CuMnFA2, CuMnFA3 and CuMnFA4 catalysts, respectively. As compared to stagnant air calcination, the flowing air produced more active catalysts for CO oxidation at a low temperature. The order of activity for various calcination conditions was as follows: FAC > SAC. The activity of the catalysts seems to be dominated both by their average oxidation numbers and the presence of different species in a catalyst surfaces. The order of activity of different types of CuMnOx catalysts for CO oxidation was as follows: CuMnFA1 > CuMnFA2 > CuMnFA3 > CuMnFA4. After the activity test, we have observed that the CuMnFA1 catalyst has a higher catalytic activity for CO oxidation, as compared to the other catalysts (Table 8).
Keeping the same precipitant while changing the precursor had a significant influence on the CO oxidation activity. The catalysts prepared by a combination of {Mn(Ac)2 + Cu(NO3)2} as precursors were considerably more active than the other catalysts. With the same precipitant, the catalysts prepared with Mn–acetate as a part of the precursor resulted in a better catalytic activity than the Mn–nitrates. A suitable combination of various chemical compounds present in the precursor resulted in the most efficient CO oxidation catalyst. The preparation of catalyst was highly influenced by their structure and activity. CO was rapidly adsorbed and occupies the active sites on the CuMnOx catalyst’s surface during the completing adsorption process.
Reactive calcination
The RC process minimized a process step by converting two-step processes into single-step process in a reactive CO–air mixture at 300 °C. It also produced CuMnRC catalysts with improved performance for CO oxidation. In the beginning, a very slow exothermic oxidation of CO over the precursor’s crystallites started causing a small rise in the local temperature, ensuing decomposition of the precursor also (Fig. 10).

Catalytic activity of CuMnFA catalysts for CO oxidation
The oxidation of CO was just initiated in reactive calcination conditions at 25 °C over all the catalyst samples and 50% conversion of CO over the CuMnRC1 catalyst was 45 °C, which was less by 10 °C, 25 °C and 40 °C over than that of CuMnRC2, CuMnRC3 and CuMnRC4 catalysts, respectively. The total oxidation temperature of CO was 80 °C for CuMnRC1 catalyst, which was less by 20 °C, 35 °C and 50 °C over than that of CuMnRC2, CuMnRC3 and CuMnRC4 catalysts, respectively. It was clear from the Table 9 and Fig. 11 that the CuMnRC1 catalyst has shown that the best catalytic activity towards CO oxidation as compared to the other catalysts. The order of activity of different types of CuMnOx catalysts for CO oxidation was as follows: CuMnRC1 > CuMnRC2 > CuMnRC3 > CuMnRC4. With the same precipitant, the catalysts prepared by Cu–Nitrate as a part of the precursor resulted in a better catalytic activity than the catalyst prepared by Cu–Acetate as a precursor.

Catalytic activity of CuMnRC catalysts for CO oxidation
The presence of Mn–Acetate and Cu–Nitrate in the CuMnRC1 precursor has shown the best catalytic activity towards CO oxidation. After the activity test, we can propose that the CuMnRC1 catalyst has a higher catalytic activity for CO oxidation as compared to the other catalysts. The CuMnRC1 catalyst has shown a promising performance for CO oxidation at a low temperature and these systems were now worthy for further investigation.
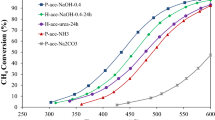
Comparison of reactive calcination with traditional calcination
The RC route was the most appropriate calcination strategy for the production of highly active CuMnOx catalyst for CO oxidation. A comparative study of CO oxidation over the (CuMnSA1, CuMnFA1 and CuMnRC1) catalysts produced under the various calcination conditions of stagnant air, flowing air and RC are shown in the Fig. 12. The calcination strategies have a drastic effect on the activity of resulting catalyst. The oxidation of CO was just initiated at 25 °C over all the catalyst samples and 50% conversion of CO over the CuMnRC1 catalyst was 45 °C, which was less by 25 °C and 45 °C over than that of CuMnFA1 and CuMnSA1 catalysts, respectively. The complete conversion of CO has occurred at 80 °C for CuMnRC1 catalyst, which was less by 40 °C and 100 °C over than that of CuMnSA1 and CuMnFA1 catalysts, respectively. The activity order for CO oxidation in the decreasing sequence was in accordance with their characterization by SEM–EDX, BET, XRD, XPS and FTIR as follows: CuMnRC1 > CuMnFA1 > CuMnSA1. The improved catalytic activity of the catalyst can be ascribed to the unique structural and textural characteristics as the smallest crystallites of CuMnRC1. The highly dispersed and specific surface area which could expose more active sites for the catalytic oxidation and relatively open-textured pores will favor the adsorption of reactants and desorption of products and thus facilitate the oxidation process. The order of activity for various calcination conditions was as follows: RC > FAC > SAC.

Activity test of CuMnOx catalysts under various calcination conditions
The partial reduction phases were provided oxygen deficient on defective structure, which also created high density for active sites in reactive calcinations process, as a result CuMnRC1 turned into the most active catalyst [14]. The highest activity of CuMnRC1 was associated with the formation of smallest crystallites authenticated by XRD observation. Using the CuMnOx catalyst, the precursors have changed the high influence activity of the resulting catalysts. Finally, it was found that the various precursors could lead to different crystalline phase formations of the CuMnOx catalysts (Table 10).
The results were confirmed by matching with the XRD, FTIR, and BET analysis. The combination of precipitant and precursor in the precipitation process plays a significant role in producing the highly active catalyst. It was postulated that during the RC method, concurrent multifarious phenomena of CO oxidation and the precursor decomposition cause a synergistic effect in the formation of oxygen-deficient catalyst structure at a low temperature. The calcination strategies influenced the physicochemical properties and catalytic performance of CuMnOx catalysts for CO oxidation [43, 44].
Stability test
The stability test of CuMnRC1 catalyst was conducted at 80 °C for oxidation of CO in a continuous running for 48 h under the earliest mentioned experimental conditions. The result revealed that practically 2% deactivation of the CuMnRC1 catalyst has occurred in the experiments’ conditions. The conversion of CO changed from 100 to 98% with the increase of time up to 48 h. The extraordinary performance of CuMnRC1 catalyst produced by RC for CO oxidation was associated with the modification in intrinsic textural, morphological characteristics such as surface area, crystallite size, and particle size of the catalyst. The main objective of this study was to evaluate the stability of CuMnRC1 catalyst as well as their importance for CO2 formation.
Conclusions
The CuMnOx catalysts have been synthesized using {Mn(Ac)2 + Cu(NO3)2}, {Mn(Ac)2 +Cu(Ac)2}, {Mn(NO3)2 + Cu(NO3)2}, {Mn(NO3)2 + Cu(AC)2} as precursor precipitated by KMnO4 solution and their catalytic activity for CO oxidation have been evaluated. The various precursors had a significant influence on the structural properties and their catalytic activity for CO oxidation. The catalyst prepared by {Mn(Ac)2 + Cu(NO3)2} as a precursor exhibited the best catalytic activity towards CO oxidation due to their high oxygen mobility. Maintaining the same precipitant, while changing the precursor causes a small change in the quantity of active sites which influence the CO oxidation activity. The calcination strategies of the precursor have a great influence on the activity of resulting catalysts. The calcination order with respect to the performance of catalysts for CO oxidation was as follows: reactive calcination > flowing air > stagnant air. The RC route was the most appropriate calcination strategy for the production of highly active CuMnRC1 catalyst for CO oxidation. It was in accordance with their characterization. The performance of this catalyst was judged by their activity, selectivity, and stability. Finally, we conclude that the choice of precursors in the catalyst is important in designing the most efficient CO oxidation catalyst.
References
Singh P, Prasad R (2014) Catalytic abatement of cold start vehicular CO emissions. Catal Ind 6(2):122–127
Dey S, Dhal GC, Prasad R, Mohan D (2016) Effect of nitrate metal (Ce, Cu, Mn and Co) precursors for the total oxidation of carbon monoxide. Resour Effic Technol 3:293–302
Cholakov GS (2010) Control of exhaust emissions from internal combustion engine vehicles. Pollut Control Technol 3:1–8
Mishra A, Prasad R (2014) Preparation and application of perovskite catalysts for diesel soot emissions control: an overview. Catal Rev Sci Eng 56:57–81
Katz M (1953) The heterogeneous oxidation of carbon monoxide. Adv Catal 5:177–216
Cai L, Guo Y, Lu A, Branton P, Li W (2012) The choice of precipitant and precursor in the co-precipitation synthesis of copper manganese oxide for maximizing carbon monoxide oxidation. J Mol Catal A Chem 360:35–41
Kramer M, Schmidt T, Stowe K, Maier WF (2006) Structural and catalytic aspects of sol–gel derived copper manganese oxides as low-temperature CO oxidation catalyst. Appl Catal A 302:257–263
Huang T, Tsai D (2003) CO oxidation behavior of copper and copper oxides. Catal Lett 87:173–178
Spivey JJ (1987) Complete catalytic oxidation of volatile organics. Am Chem Soc 26:2165–2180
Zhou Y, Wang Z, Liu C (2014) Perspective on CO oxidation over Pd-based catalysts. Catal Sci Technol 5:69–81
Wang S, Zheng X, Wang X, Wang S, Zhang S, Yu L, Huang W, Wu S (2005) Comparison of CuO/Ce0.8Zr0.2O2 and CuO/CeO2 catalysts for low-temperature CO oxidation. Catal Lett 105(3–4):163–168
Santra AK, Goodman DW (2002) Catalytic oxidation of CO by platinum group metals: from ultrahigh vacuum to elevated pressures. Electrochim Acta 47:3595–3609
Solsona B, Hutchings GJ, Garcia T, Taylor SH (2004) Improvement of the catalytic performance of CuMnOx catalysts for CO oxidation by the addition of Au. New J Chem 28:708–711
Hutchings GJ, Mirzaei AA, Joyner RW, Siddiqui MRH, Taylor SH (1996) Ambient temperature CO oxidation using copper manganese oxide catalysts prepared by co-precipitation: effect of ageing on catalyst performance. Catal Lett 42:21–24
Jones C, Cole KJ, Taylor SH, Crudace MJ, Hutchings GJ (2009) Copper manganese oxide catalysts for ambient temperature carbon monoxide oxidation: effect of calcination on activity. J Mol Catal A Chem 305:121–124
Tanaka Y, Utaka T, Kikuchi R, Takeguchi T, Sasaki K, Eguchi K (2003) Water gas shift reaction for the reformed fuels over Cu/MnO catalysts prepared via spinel-type oxide. J Catal 215:271–278
Peng CT, Lia HK, Liaw BJ, Chen YZ (2011) Removal of CO in excess hydrogen over CuO/Ce1−XMnxO2 catalysts. Chem Eng J 172:452–458
Lee J, Kim H, Lee H, Jang S, Chang JH (2016) Highly efficient elimination of carbon monoxide with binary copper-manganese oxide contained ordered nanoporous silicas. Nanoscale Res Lett 11:2–6
Dey S, Dhal GC, Prasad R, Mohan D (2017) Effects of doping on the performance of CuMnOx Catalyst for CO oxidation. Bull Chem React Eng Catal 12(3):1–14
Qian K, Qian Z, Hua Q, Jiang Z, Huang W (2013) Structure activity relationship of CuO/MnO2 catalysts in CO oxidation. Appl Surf Sci 273:357–363
Irawan RB, Purwanto P, Hadiyanto H (2015) Optimum design of manganese-coated copper catalytic converter to reduce carbon monoxide emissions on gasoline motors. Int Conf Trop Coast Reg Eco-Dev 23:86–92
Marinoiu A, Raceanu M, Cobzaru C, Teodorescu C, Marinescu D, Soare A, Varlam M (2014) Low temperature CO retention using hopcalite catalyst for fuel cell applications. React Kinet Mech Catal 112:37–50
Hu Y, Dong L, Wang J, Ding W, Chen Y (2000) Activities of supported copper oxide catalysts in the NO + CO reaction at low temperatures. J Mol Catal A Chem 162:307–316
Liu Y, Guo Y, Peng H, Xu X, Wu Y, Peng C, Zhang N, Wang X (2016) Modifying Hopcalite catalyst by SnO2 addition: an effective way to improve its moisture tolerance and activity for low temperature CO oxidation. Appl Catal A 525:204–214
Mokhtar M, Basahel SN, Al-Angary YO (2010) Nanosized spinel oxide catalysts for CO-oxidation prepared via CoMnMgAl quaternary hydrotalcite route. J Alloy Compd 493:376–384
Choi K, Lee D, Kim H, Yoon Y, Park C, Kim YH (2016) Reaction characteristics of precious-metal-free ternary Mn − Cu − M (M = Ce Co, Cr, and Fe) oxide catalysts for low-temperature CO oxidation. Ind Eng Chem Res 55:4443–4450
Larsson P, Andersson A (2000) Oxides of copper, ceria promoted copper, manganese and copper manganese on Al2O3 for the combustion of CO, ethyl acetate and ethanol. Appl Catal B 24:175–192
Dey S, Dhal GC, Mohan D, Prasad R (2017) Kinetics of catalytic oxidation of carbon monoxide over CuMnAgOx Catalyst. Mater Discov 8:18–25
Dey S, Dhal GC, Mohan D, Prasad R (2017) Study of Hopcalite (CuMnOx) catalysts prepared through a novel route for the oxidation of carbon monoxide at low temperature. Bull Chem React Eng Catal 12(3):393–407
Njagi EC, Chen C, Genuino H, Galindo H, Huang H, Suib SL (2010) Total oxidation of CO at ambient temperature using copper manganese oxide catalysts prepared by a redox method. Appl Catal A 99:103–110
Dey S, Dhal GC, Mohan D, Prasad R (2017) Characterization and activity of CuMnOx/γ-Al2O3 catalyst for oxidation of carbon monoxide. Mater Discov 8:26–34
Dey S, Dhal GC, Prasad R, Mohan D (2016) Total oxidation of CO by CuMnOx catalyst at a low temperature. Int J Sci Eng Res 7(10):1730–1737
Zhang XD, Hou FL, Li HX, Yang Y, Wang YX, Liu N, Yang YQ (2018) A strawsheave-like metal organic framework Ce-BTC derivative containing high specific surface area for improving the catalytic activity of CO oxidation reaction. Microporous Mesoporous Mater 259:211–219
Dey S, Dhal GC, Mohan D, Prasad R (2017) Effect of preparation conditions on the catalytic activity of CuMnOx catalysts for CO oxidation. Bull Chem React Eng Catal 12(3):437–451
Zhang XD, Li HX, Yang Y, Zhang TT, Wen X, Liu N, Wang DJ (2017) Facile synthesis of new efficient Cu/MnO2 catalysts from used battery for CO oxidation. J Environ Chem Eng 5:5179–5186
Yang YQ, Dong H, Wang Y, Wang YX, Liu N, Wang DJ, Zhang XD (2017) A facile synthesis for porous CuO/Cu2O composites derived from MOFs and their superior catalytic performance for CO oxidation. Inorg Chem Commun 86:74–77
Zhang XD, Li HX, Hou FL, Yang Y, Dong H, Liu N, Wang YX, Cui LF (2017) Synthesis of highly efficient Mn2O3 catalysts for CO oxidation derived from Mn-MIL-100. Appl Surf Sci 411:27–33
Zhang XD, Hou FL, Yang Y, Wang YX, Liu N, Chen D, Yang YQ (2017) A facile synthesis for cauliflower like CeO2 catalysts from Ce-BTC precursor and their catalytic performance for CO oxidation. Appl Surf Sci 423:771–779
Cui LF, Zhao D, Yang Y, Wang YX, Zhang XD (2017) Synthesis of highly efficient α-Fe2O3 catalysts for CO oxidation derived from MIL-100(Fe). J Solid State Chem 247:168–172
Zhang XD, Dong H, Wang Y, Liu N, Zuo YH, Cui LF (2016) Study of catalytic activity at the Ag/Al-SBA-15 catalysts for CO oxidation and selective CO oxidation. Chem Eng J 283:1097–1107
Dey S, Dhal GC, Mohan D, Prasad R, Gupta RN (2018) Cobalt doped CuMnOx catalysts for the preferential oxidation of carbon monoxide. Appl Surf Sci 441:303–316
Yang YQ, Hou FL, Li HX, Liu N, Wang Y, Zhang XD (2017) Facile synthesis of Ag/KIT-6 catalyst via a simple one pot method and application in the CO oxidation. J Porous Mater 24:1661–1665
Dey S, Dhal GC, Mohan D, Prasad R (2018) Low-temperature complete oxidation of CO over various manganese oxide catalyst. Atmos Pollut Res 9:755–763
Dey S, Dhal GC, Mohan D, Prasad R (2018) Copper based mixed oxide catalysts (CuMnCe, CuMnCo and CuCeZr) for the oxidation of CO at low temperature. Mater Discov 10:1–14
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dey, S., Dhal, G.C., Mohan, D. et al. The choice of precursors in the synthesizing of CuMnOx catalysts for maximizing CO oxidation. Int J Ind Chem 9, 199–214 (2018). https://doi.org/10.1007/s40090-018-0150-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40090-018-0150-7