
Abstract
Lately we have proposed an atomic polarizability model, viz. \(\alpha \propto \left({r}^{3}/{Z}_{\mathrm{eff}}{e}^{2}\right)\), through an empirical approach. As the results obtained using the model were remarkable, we have tried to explore the efficacy of this polarizability model by using four different types of radii for 96 atoms invoking a regression analysis. Further, we have performed a study on molecules by employing additivity property. Although the results are similar in the case of atoms, two of the four radii-based polarizability sets perform better when molecules are considered. In addition, the molecular polarizability is computed for a variety of anaesthetics due to its significance in biochemical interactions. A significant correlation is obtained between the computed and the published data, corroborating the efficacy of polarizability model in the prediction of biological mechanisms. The polarizability model is revealed to be conceptually rigorous even when different types of radii are used, so it can be satisfactorily employed for real-field applications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Evaluation of the polarizability is currently a vigorously expanding field of study. Extensive theoretical, as well as experimental, works are being carried out for its computation in case of atoms, molecules, ions and clusters. By definition, the polarizability is the ease of distortion of an electronic cloud corresponding to a species. Several reviews that have been published on the topic report a wide range of theoretical and experimental approaches for the calculation of polarizabilities [1, 2]. Indeed, the relationship of polarizability to other descriptors has been explored by numerous scientists [3,4,5,6,7]. The effect of polarizability on the nucleus of an atom [8] and the mechanical properties of macro- and nano-dimensional organic cocrystals has been studied, as well [9], and the polarizability has been revealed to be of immense importance in understanding and predicting the nature of chemical–biological interactions [10, 11].
Due to the wide applicability of the polarizability in diverse realms, a polarizability model relying on the mutual effect of the effective atomic nuclear charge (Zeff) and atomic radius (r) was recently proposed by our group [12]. The model is based on an empirical approach and follows all the criteria of a descriptor.
The present work is a quest for assessing the potential of the previously proposed model and exploring its application in molecular polarizability calculations invoking the additivity concept. With an intention of testing the efficacy of the model in different scenarios, we employ different kinds of radii to compute four sets of polarizability. Further, we have tried to study the role of our computed polarizabilities in some biochemical species.
The radius of an atom is a size descriptor. It is a valuable quantity that supports understanding of many physico-chemical properties of different species. It also plays an important role in interpreting various biochemical processes. However, the concept of the radius of an atom is still unclear. Theoretical calculations of atomic and ionic sizes have evolved from an empirical model and have reached up to Self-Consistent Field theory [3, 13,14,15,16,17,18,19,20,21]. A number of terms, such as the atomic radii, van der Waals radii, metallic radii, covalent radii, ionic radii, and absolute radii, and estimation approaches such as empirical, crystallographic, self-consistent field (SCF)-based, orbital-density based, etc., are known to exist for this single property [22,23,24]. Each of these radii and approaches has some advantages and disadvantages over the others. For instance, covalent radii help in providing information regarding the existence of a chemical bond, but cannot provide a fixed value as it is affected by its surroundings. This radius can be valuable when molecules with different types of bonds, viz. single, double and triple, come into the picture. Similarly, an absolute radius can offer a universal value for each atom, but it may lack in explaining the type of bond in some cases. This radius works best in situations where transferability is highly crucial, such as clusters, nanoparticles, etc. Moreover, an ionic radius keeps changing even if a single electron is gained or lost. Ionic radii are basically useful in understanding the structure of ionic crystals and solvated ions in liquids. An empirical relationship is popular in terms of the minimum requirement for computational resources, whereas SCF has inherent importance in its accurate prediction of atomic and ionic size. Thus, until now, no fixed or universal benchmark for the radius of an atom/ion/molecule has existed. Considering this difficulty, we have tested the validity of our proposed polarizability model for four different kinds of radii.
Numerous studies have utilized chemical reactivity descriptors to explain the mechanisms of chemical–biological interactions [6, 11, 25,26,27,28,29,30,31,32,33,34]. One such important descriptor is polarizability. In a study by Agin and others [25], polarizability was used to analyse the potential of various chemicals that hindered action in a frog’s muscle. A similar study on frog muscle was performed by Kamlet et al. using several drugs [26]. Hahin and his group studied the effect of alcohols on the nerves of frogs through polarizability [27]. Many phenyl alkane p-ω-bis(trialkylammonium) compounds were examined by Wien and Mason, who employed polarizability to study their pharmacological activities [28]. Nishimura et al. studied the mechanism of reduction in the action potential in the central nerve cord by means of polarizability [29]. An analysis was made by Tandon et al. to understand the role and the mechanism of polarizability in ligand–substrate biological interactions [11]. It is evident that several biological processes depend on the circulation and dispersal of chemicals within a living organism and such activities are believed to be significantly governed by the polarizability along with some other electronic effects. Thus, we have tried to demonstrate the potential of the polarizability model in providing molecular polarizabilities that can be employed to study electronic interactions in biochemical systems.
2 Method of computation
The polarizability and the radius of an atom, as described above, are important reactivity descriptors [35,36,37,38]. Thus, we have used four different sets of radii to explore the efficacy of the polarizability model as proposed by our group previously [12]. Based on the previously proposed model, the polarizability (α) is proportional to the ratio of the cube of atomic radius (r) and the product of effective nuclear charge (Zeff) and the square of electronic charge (e) as follows (Eq. 1),
Here a and b represent regression parameters. In the present study, we have employed Clementi et al.’s SCF-based radii [39, 40], Desclaux’s orbital density-based radii [41], Cordero et al.’s covalent radii [42] and Tandon et al.’s absolute radii [24] to compute the polarizability by using Eq. (2). The reason for selecting these radii is to check the suitability of a variety of radii obtained using diverse methods in the computation of the polarizability.
The polarizability computations are performed for the atoms of 96 elements in the periodic table through a regression approach invoking Eq. (2). For the analysis, the radius and the effective nuclear charge serve as independent variables, whereas polarizability acts as a dependent variable. Reference polarizabilities are obtained from Schwerdtfeger and Nagle’s work [43]. Radii are taken from Clementi et al.’s [39, 40], Desclaux’s [41], Cordero et al.’s [42] and Tandon et al.’s [24] work while Ghosh and Biswas’s effective nuclear charge [15] is used for this purpose. All the radii as mentioned in the report are in units of Å. The procedure involves evaluation of the parameters a and b by using linear regression, performed period-wise, followed by a computation of the atomic polarizability based on the obtained data. The four types of computed polarizabilities are compared with one another and with the Schwerdtfeger and Nagle’s [43] polarizabilities in order to test their accuracy and find the most effective amongst all. The relationship of the ionization energy to the polarizability has also been demonstrated by a number of scientists earlier [7, 23, 43,44,45,46,47,48,49,50]. Accordingly, we have made an attempt to find the correlation between these two properties for each period based on an empirical power relationship [51].
We have also tested the potential of using these polarizabilities for molecules. For this purpose, we have calculated the molecular polarizabilities by using all four computed atomic polarizability values for some simple molecules. The molecular value is calculated employing the property of additivity [52,53,54] (see Eq. 3). As per the property, “the summation of the polarizabilities of all the isolated atoms present in a system gives an approximate value of the static molecular polarizability (αm)”:
The obtained molecular polarizabilities are contrasted against the published theoretical polarizabilities of van Duijnen and Swart [55].
Because polarizability influences the reactivity and the mechanism of various biological molecules, drugs, anaesthetics, etc., as evidenced by the literature [11, 25,26,27,28,29], we have made an effort to study the effect of the computed polarizabilities on some anaesthetics, as well. We have calculated the molecular polarizabilities by using our computed polarizability data based on Desclaux’s, Cordero et al.’s and Tandon et al.’s radii, for some anaesthetics that cause nerve inhibition due to their intake leading to an excitability block in a frog’s sartorius muscle. In order to verify the validity of the computed values, we performed a comparison with the molecular values published by Tandon et al. [11].
3 Results and discussion
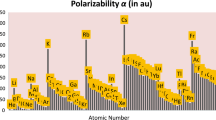
Atomic polarizabilities computed by employing four kinds of radii, viz. Clementi–Raimondi [39] and Clementi et al. radii [40] (αC), Desclaux radii [41] (αD), Cordero et al. radii [42] (αCo) and Tandon et al. [24] (αT), are presented in Table 1 for 96 elements. The magnitude of these polarizabilities is observed to vary from one another; however, such small differences are acceptable. A look at Fig. 1 shows that each polarizability exhibits a similar periodicity trend. This figure further corroborates the fact that the magnitudes of all the values are nearly the same. These results suggest that the model functions suitably for most of the radii when considering atoms. In spite of the radii being calculated through very different approaches and based on different principles, these provide acceptable results for atomic polarizabilities.

Comparative plot of polarizabilities computed using Clementi–Raimondi and Clementi et al. radii (αC/Å3), Desclaux radii (αD/Å3), Cordero et al. radii (αCo/Å3), Tandon et al. radii (αT/Å3), and Schwerdtfeger and Nagle’s [43] polarizability (α/Å3) values
We have also correlated all the computed polarizabilities with ionization energy for each period. The empirical power relationships of all the polarizability sets (αC, αD, αCo and αT) to the ionization energy reveal nice correlations (r2 > 0.8) between the descriptors, which suggests the presence of a useful quantitative association between these sets of polarizability and ionization energy.
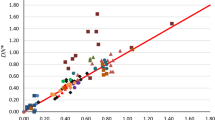
The efficacy of these polarizabilities is also confirmed for molecules. We have used αC, αD, αCo and αT for calculating the molecular polarizabilities of some simple molecules by invoking the additive property. We observed that the molecular polarizabilities calculated using αCo and αT present excellent correlations with the corresponding theoretical values. However, molecular data determined using αC and αD provide comparatively low correlations with the published theoretical data [55]. The order of decreasing correlation between the theoretical and the computed sets of molecular polarizability is αmCo (r2 = 0.9453) > αmT (r2 = 0.8934) > αmC (r2 = 0.7899) > αmD (r2 = 0.7326). Table 2 lists the theoretical and the calculated molecular polarizabilities for the molecules under study. The molecular polarizability values calculated using αCo and αT are noted to be more accurate than the values calculated using the other two parameters using the same model. We believe that the variation in the four sets of computed molecular values may be due to the presence of relativistic effect in the polarizability data calculated by Cordero et al. [42] and Tandon et al. radii [24].
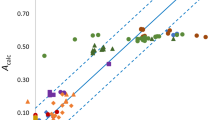
The molecular polarizability results are also validated in the case of anaesthetics. Because the polarizability is believed to be associated with the mechanism of anaesthetic action [11], which occurs through an electrically excitable membrane, verifying the results in the case of such molecules is also important. The molecules for the study were selected so that heterogeneity is incorporated in the chemical organization. Table 3 presents the three sets of molecular polarizabilities calculated using the computed Desclaux radii-based polarizability, Cordero et al. radii-based polarizability and Tandon et al. radii-based polarizability along with the theoretical molecular polarizabilities [11] for some anaesthetics. As is evident from the table, the molecular polarizability values for each set are very close to their theoretical counterparts. The correlation plots in Fig. 2 signify an excellent relationship between these computed data and the theoretical data. The same trend is observed again for the theoretical versus the computed sets of molecular polarizability: αmCo (r2 = 0.9987) > αmT (r2 = 0.9969) > αmD (r2 = 0.9968). This further confirms the suitability of using Cordero et al.’s radii [42] followed by Tandon et al.’s radii [24] for computational purpose for the previously proposed polarizability model [12]. The much smaller variation in these correlations, as well as in the magnitude of polarizability, also indicates the efficacy of the model in different scenarios. This suggests that the computed polarizability model values can be employed to study the mechanism of anaesthetic action solely or in combination with other descriptors.

Correlation plots of theoretical molecular polarizabilities (αm/Å3) [11] versus computed molecular polarizabilities calculated using a Desclaux radii-based polarizability (αmD/Å3); b Cordero et al. radii-based polarizability (αmCo/Å3), and c Tandon et al. radii-based polarizability (αmT/Å3) for some anaesthetics
4 Conclusion
The present work explores the potential of a polarizability model, viz. \(\alpha \propto \left({r}^{3}/{Z}_{\mathrm{eff}}{e}^{2}\right)\), proposed by our group previously [12]. The efficacy is checked using four different sets of radii for polarizability computations. Suitable similarity in magnitude is observed amongst all the computed polarizabilities. Further molecular polarizabilities are computed invoking the property of additivity, and the results show a valuable correlation with the published data. Both these results indicate the accuracy of the model. However, we note that the Cordero et al. [42] radius-based and Tandon et al. [24] radius-based atomic polarizabilities provide superior results in the case of molecules. Molecular data are also calculated for a range of anaesthetics owing to their relevance in chemical–biological interactions. Obtained results reveal significant correlation with the published data, validating the potential for the polarizability model to be used in predicting biological mechanisms as well. Hence, clearly, the polarizability model is conceptually sound and can be satisfactorily employed for real-field applications.
References
K.D. Bonin, M.A. Kadar-Kallen, J. Mod. Phys. B 8, 3313 (1994)
J.A. Mitroy, M.S. Safronova, C.W. Clark, J. Phys. B 43, 202001 (2010)
J.K. Nagle, J. Am. Chem. Soc. 11, 4741 (1990)
D.C. Ghosh, K. Gupta, J. Theor. Comput. Chem. 5, 895 (2006)
J. Wang, P. Cieplak, J. Li, T. Hou, R. Luo, Y. Duan, J. Phys. Chem. B 115, 3091 (2011)
H. Tandon, T. Chakraborty, V. Suhag, Int. J. Quant. Struct. Prop. Relationsh. 4, 99 (2019)
H. Tandon, T. Chakraborty, V. Suhag, J. Math. Chem. 57, 2142 (2019)
J.N. Orce, Int. J. Mod. Phys. E 29, 2030002 (2020)
T.P. Rupasinghe, K.M. Hutchins, B.S. Bandaranayake, S. Ghorai, C. Karunatilake, D.K. Bučar, D.C. Swenson, M.A. Arnold, L.R. MacGillivray, A.V. Tivanski, J. Am. Chem. Soc. 137, 12768 (2015)
C. Hansch, W.E. Steinmetz, A.J. Leo, S.B. Mekapati, A. Kurup, D. Hoekman, J. Chem. Inform. Comput. Sci. 43, 120 (2003)
H. Tandon, P. Ranjan, T. Chakraborty, V. Suhag, Mol. Divers. 25, 249 (2021)
S. Choudhary, P. Ranjan, T. Chakraborty, J. Chem. Res. 44, 227 (2020)
J.C. Slater, J. Chem. Phys. 41, 3199 (1964)
J.C. Slater, Quantum Theory of Molecules and Solids: Symmetry and Energy Bands in Crystals, vol. 2 (McGraw-Hill, New York, 1963).
D.C. Ghosh, R. Biswas, Int. J. Mol. Sci. 3, 87 (2002)
M.V. Putz, N. Russo, E. Sicilia, J. Phys. Chem. A 107, 5461 (2003)
P. Pyykkö, S. Riedel, M. Patzschke, Chem. Eur. J. 11, 3511 (2005)
H. Tandon, T. Chakraborty, V. Suhag, Mol. Phys. 119, e1820594 (2021)
P. Szarek, A. Chlebicki, W. Grochala, J. Phys. Chem. A 123, 682 (2019)
M. Rahm, R. Hoffmann, N.W. Ashcroft, Chem. Eur. J. 22, 14625 (2016)
J. Gebhardt, A.M. Rappe, Comput. Phys. Commun. 237, 238 (2019)
J.E. Huheey, E.A. Keiter, R.L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity (Addison-Wesley, New York, 1993).
P. Politzer, P. Jin, J.S. Murray, J. Chem. Phys. 117, 8197 (2002)
H. Tandon, P. Ranjan, T. Chakraborty, V. Suhag, J. Math. Chem. 58, 1025 (2020)
D. Agin, L. Hersh, D. Holtzman, Proc. Natl. Acad. Sci. USA. 53, 952 (1965)
M.J. Kamlet, R.M. Doherty, M.H. Abraham, R.W. Taft, Quant. Struct. Act. Relatsh. 7, 71 (1988)
R. Hahin, A. Kondratiev, J. Membr. Biol. 180, 137 (2001)
R. Wien, D.F.J. Mason, Br. J. Pharmacol. Chemother. 8, 306 (1953)
K. Nishimura, M. Ohoka, T. Fujita, Pestic. Biochem. Physiol. 28, 257 (1987)
A.G. Mercader, A.B. Pomilio, Eur. J. Med. Chem. 45, 1724 (2010)
L. Bober, P. Kawczak, T. Baczek, Lett. Drug Des. Discov. 9, 595 (2012)
P. Kawczak, L. Bober, T. Baczek, Curr. Pharm. Anal. 10, 255 (2014)
K. Roy, S. Kar, R.N. Das, Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment (Academic press, New York, 2015).
H. Tandon, T. Chakraborty, V. Suhag, J. Mol. Model. 25, 303 (2019)
D.S. Sabirov, R.G. Bulgakov, Fuller. Nanotub. Carbon Nanostruct. 18, 455 (2010)
W. Carruthers, I. Coldham, Modern Methods of Organic Synthesis (Cambridge University Press, Cambridge, 2004).
T.H. Lowry, K.S. Richardson, Mechanism and Theory in Organic Chemistry (Harper & Row, New York, 1987).
F.A. Carey, R.J. Sundberg, Advanced Organic Chemistry Part a: Structure and Mechanisms (Springer, New York, 2007).
E. Clementi, D.L. Raimondi, J. Chem. Phys. 38, 2686 (1963)
E. Clementi, D.L. Raimondi, W.P. Reinhardt, J. Chem. Phys. 47, 1300 (1967)
J.P. Desclaux, At. Data Nucl. Data Tables 12, 311 (1973)
B. Cordero, V. Gómez, A.E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán, S. Alvarez, Dalton Trans. (2008). https://doi.org/10.1039/b801115j
P. Schwerdtfeger, J.K. Nagle, Mol. Phys. 117, 1200 (2019)
I.K. Dmitrieva, G.I. Plindov, Phys. Scr. 27, 402 (1983)
P. Politzer, J.S. Murray, M.E. Grice, T. Brinck, S. Ranganathan, J. Chem. Phys. 95, 6699 (1991)
B. Fricke, J. Chem. Phys. 84, 862 (1986)
I.K. Dmitrieva, G.I. Plindov, J. Appl. Spectrosc. 44, 4 (1986)
H.J. Bohórquez, R.J. Boyd, Chem. Phys. Lett. 480, 127 (2009)
R.L. DeKock, J.R. Strikwerda, E.X. Yu, Chem. Phys. Lett. 547, 120 (2012)
U. Hohm, A.J. Thakkar, J. Phys. Chem. A 116, 697 (2012)
A. Kramida, Y. Ralchenko, J. Reader, NIST ASD Team, NIST Atomic Spectra Database, Version 5.6.1 (National Institute of Standards and Technology, Gaithersburg, MD, 2018). https://physics.nist.gov/asd. Accessed 10 Dec 2020
P. Politzer, J.S. Murray, F.A. Bulat, J. Mol. Model. 16, 1731 (2010)
Y.K. Kang, M.S. Jhon, Theor. Chim. Acta 61, 41 (1982)
K.J. Miller, J. Am. Chem. Soc. 112, 8533 (1990)
P.T. van Duijnen, M. Swart, J. Phys. Chem. A 102, 2399 (1998)
Acknowledgements
Dr. Tanmoy Chakraborty is thankful to Sharda University, and Dr. Hiteshi Tandon and Dr. Shalini Chaudhary are thankful to Manipal University Jaipur, for providing computational resources and a research facility.
Author information
Authors and Affiliations
Contributions
SC: resources, formal analysis, writing—original draft. HT: conceptualization, methodology, formal analysis, investigation, validation, writing—original draft, visualization. TC: conceptualization, supervision, writing—review and editing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chaudhary, S., Tandon, H. & Chakraborty, T. A quest for effective polarizability as a function of the radii. J. Korean Phys. Soc. 78, 1101–1108 (2021). https://doi.org/10.1007/s40042-021-00130-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40042-021-00130-1