
Abstract
Marek’s disease (MD) is a lymphoproliferative and neuropathic disease of domestic fowl caused by an oncogenic Gallid Herpesvirus-2 (GaHV-2), also known as Marek’s Disease Virus serotype 1 (MDV-1). A total of forty (n = 40) liver samples of 14 outbreaks at poultry farm suspected for MD were investigated. Collected samples were processed for classical as well as for molecular diagnosis and further characterization of pathogens for the control of the disease. The Meq gene-based real-time PCR, nested PCR and virus isolation in CEF were performed. Out of 40 biological samples, 80% (32/40) samples were detected positive for MDV-1 by real-time PCR, 67.5% (27/40) by nested PCR and only 25.9% (7/27) by virus isolation in CEF. The nucleotide and deduced amino acid sequence data analysis of Meq gene revealed the presence of virulent MDV-1 strain in the majority of the samples of outbreaks. The mild virulent strain was also confirmed in only one field samples with maximum sequence identity with vaccine strain from the Netherlands (CVI988) and China (814). Present study conclude that the virulent strain of MDV-1 is circulating in Haryana state of India and responsible for morbidity and mortality among vaccinated poultry birds.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Marek’s disease (MD) is caused by the cell-associated α-herpesvirus, i.e., Gallid herpesvirus 2 (GaHV-2) which belong to family—Herpesviridae, subfamily—Alphaherpesvirinae and genus—Mardivirus. It is well-known disease of chickens having lymphoproliferative signs. MD is responsible for causing serious economic losses to the poultry industry. There are three serotypes of MD virus which share common antigen [1]. These serotypes include type-1 containing oncogenic MDV-1, type -2 containing non-oncogenic MDV-2 and type-3 containing herpesvirus of turkey (HVT) [2]. Commercially available HVT vaccines have well controlled the disease. However, the proximity of large flocks of chickens and varied level of immune and health status of birds has led to emergence of diversity in MDV-1. Due to extensive vaccination of flocks against MDV-1, the immune evading strains are evolving day by day which have great genetic diversity and increased virulence for host [2, 3]. The estimated economic burden to poultry industry of MD may be nearly about US $ 1 to 2 billion annually [4].
MDV-1 infects respiratory tract of birds mainly via inhalation route. Virus particles inoculated in lungs start replicating in B and T lymphocytes resulting in initial acute cytolytic phase in lymphyocytes followed by immuno-suppression to host [5, 6]. After 6 to 7 day post-infection, the virus genome gets integrated into the genome of CD4 + T lymphocytes without any detectable expression of the potential antigenic proteins resulting in progression of latent stage of virus. This latent phase allows immune evasion by the virus which results in lymphomatous lesions in the visceral organs, peripheral nerves, and skin. The virus fully replicates only in feather follicle epithelium which act as primary source of infectious viral particles to other susceptible birds during sloughing off the wing in air [7].
The genome of MDV-1 consists of more than 200 genes. One of such gene named meq gene which has been found associated with oncogenicity. The Meq protein is the most important viral protein leading to herpesvirus-induced tumorigenicity [8]. Meq protein is a 339 amino acid protein having C-terminal trans-activation domain and N-terminal basic region leucine zipper (bZIP) domain [9]. The bZIP domain includes two stretches of basic residues (basic regions 1 and 2, i.e., BR1 and BR2) and a leucine zipper. The trans-activation domain contains 2.5 proline-rich repeats (PRRs), which has several SH3-binding motifs. Due to diverse nature of this protein, Meq is defined as a key virulence factor in poultry herpesvirus [10]. However, oncogene which contributes toward enhanced virulence of MDV-1 was remaining uncharacterized during the past studies. The Meq gene has the greatest possibility toward oncogenicity and pathogenicity of virus.
India is also a sufferer of huge economic loss of approximately 40 million Indian rupees due to MD outbreaks in vaccinated poultry flocks [11]. Several outbreaks of MDV-1 have been reported from vaccinated poultry flocks in India. In Karnataka state of India, outbreak was reported in vaccinated commercial layer flocks in mining area where huge amount of mine dust and soil cause immuno-suppression leading to MDV-1 infection [12]. The agar gel immunoprecipitation test (AGPT) had also confirmed the outbreak of MDV-1 in Mizoram state in India among poultry flocks [13]. The meq gene-based modern techniques such as PCR and nucleic acid sequencing and classical histopathology-based techniques were used for molecular characterization of Indian strains of MDV-1 from several outbreaks in Haryana [14], South India [15, 16] and North-east India [17].
The molecular diagnosis and characterization of pathogens are important for control of the disease; hence, present study was planned to identify and characterize the circulating MDV-1 strain among the poultry birds.
Material and Methods
Sample Collection
A total of 14 poultry outbreaks suspected for Marek’s disease on the basis of clinical signs were attended during the period of one year, and a total of forty (n = 40) liver samples were collected from suspected cases of MD particularly from 18 to 26-week-old vaccinated chicken flock from Hisar and its adjoining districts of Haryana. These samples were collected in 50% buffered glycerol and stored at −20 °C for further downstream processing.
Extraction of Nucleic Acid
The collected tissue samples were triturated with the help of pestle and mortar. Approximately 80–100 mg triturated tissue sample was resuspended in 400 μl of tissue lysis buffer (0.5 mol/l Tris HCl, 0.5 mol/l EDTA, 2% SDS) and 200 μg proteinase K and incubated at 55ºC for 1 h. After this, an equal volume of phenol:chloroform:isoamylalcohol (25:24:1 v/v) was added to the lysate and centrifuged. The aqueous phase was separated, and DNA was precipitated with double the volume of absolute ethanol after overnight incubation at −20 °C and centrifuged at 10,000 × g and 15–20 °C for 20 min. The pellet was washed with 70% ethanol, air dried and dissolved in 30 μL of nuclease-free water for further use.
Real-time PCR (qPCR)
All the extracted DNA samples were screened using TaqMan probe-based qPCR. The published Meq gene-specific primers and probe were used for the assay [18] (Table 1). The dual dye-labeled HPLC purified probe with 6-carboxyfluorescein (6-FAM) at 5' end and carboxytetramethylrhodamine (TAMRA) at 3' end was used. The amplification and detection were performed at real-time PCR machine (ABI 7500 standard) with the TaqMan Universal PCR Master Mix (Applied Biosystems). The real-time PCR reaction was optimized in 25 μl reaction containing 0.5 pmol of forward primer, 0.5 pmol of reverse primer and 0.25 pmol of hydrolysis probe. Cycling conditions used were 50 °C for 2 min, initial denaturation at 95 °C for 2 min followed by 40 cycles of denaturation at 95 °C for 15 s followed by annealing and extension at 60 °C for 1 min.
Nested PCR (nPCR)
The Meq gene-specific nPCR was also used for detection of MDV-1 [19] (Table 1) and generation of amplicons for sequencing. The first outer set of primer generated 1,062-bp Meq or a 1,242-bp L-Meq amplicons. The PCR amplification reaction was carried out using 3 μL (20 ng/μL) DNA as template in a total volume of 25 μL containing 12.5 μL of Top taq master mix (Qiagen, the USA), 1 μL of 10 pmol/l of each of the two primers, and 7.5 μL of nuclease-free water. Only 1.0μL of first PCR was used as template for the second round of PCR of 25 μL reaction mixture containing 0.5 μM of each primer to amplify a 583-bp meq or 763-bp L-meq fragment. The thermal cycling parameters for both sets of PCR were same except annealing step as initial denaturation at 94ºC for 4 min, 35 cycles of denaturation at 94ºC for 1 min, annealing at 56ºC for 1 min, elongation at 72ºC for 1 min, final elongation at 72ºC for 10 min and hold at 4ºC for 5 min. The second or nested set of PCR was performed at the annealing temperature of 60ºC for 30 s. The amplicons generated were separated on ethidium bromide (0.5μ/ml) containing agarose gels (1.5%) and visualized under gel documentation system.
Virus Isolation in Cell Culture
The primary cell culture of chicken embryo fibroblast (CEF) was prepared and used for virus isolation. Briefly, 10–12 days old eggs were used for preparation of chicken embryo fibroblast culture. The cells were disrupted for muscular mass and further trypsinized for separation. The CEF monolayer was prepared and incubated at 37 °C for 24 h. After this, triturated tissue extract was used for infection of cells. Cytopathic effect (CPE) was observed till 6th day of post-inoculation of virus.
Sequencing and Phylogenetic Analysis
nPCR amplified products were gel purified using QIAquick Gel Extraction Kit (Qiagen, the USA) and sequenced in Automated genetic analyzer (ABI 3130XL) using BigDye™ terminator v3.1 cycle sequencing kit (Applied Biosystems, the USA). The sequence data were assembled using BioEdit v7.2.5 program and subjected to BLASTn and ClustalW analysis for comparison with other available Meq gene sequences in Genbank. The contig sequences were used for generation of deduced amino acid sequences and nucleotide as well as deduced amino acid sequence identities was calculated using BioEdit v7.2.5 program [20]. Amino acid sequences were aligned with similar sequences from GenBank (Table 2), and phylogenetic analysis was performed using MEGA X program [21].
Results and Discussion
qPCR and nPCR
Of the 40 suspected liver samples, 32 were found positive in TaqMan probe-based qPCR with Ct value ranged from 17–36 (Fig. 1). Same samples were also screened using nPCR, and only 27 samples were found positive and yielded correct sized amplicons of 583 bp in 26 samples and 763 bp of L-meq in one sample (Fig. 2). Moreover, the real-time qPCR detected 80% (32/40) sample as positive for MDV-1 with Meq gene-specific primer and probe. However, 67.5% samples were found positive in nPCR along with one long electropherotype. The details of the result of qPCR and nPCR are given in Table 3. In previous study, meq gene specific, SYBR Green-based real-time PCR was developed to detect and quantify the MDV-1 from feather follicle which was found to be 2.5–10 times more sensitive than the conventional PCR assay [22, 23]. Similarly, in other study the meq gene-based real-time PCR was found 10–100 times more sensitive than conventional PCR in diagnosis of MDV-1 [24]. Thus, real-time PCR assay may be used as a reliable assay for monitoring the MDV-1 infected birds. The real-time PCR assay can also be used for both the epidemiological survey as well as diagnostic purpose as they have capability to differentiate and quantitate the vaccine and pathogenic strain of MDV-1 [10, 25]. Previous study had shown that some of the strains of MDV-1 such as CVI988/R6 and JM possess L-meq genes. It is well established that the L-meq gene is displayed in MDV subpopulation only during the latent phase of infection. Thus, it can be hypothesized that L-meq gene might have important role in maintenance of MDV latency in host cells [26, 27].

qPCR Log amplification curve of Meq gene of MDV-1

Agarose gel electrophoresis of amplification of meq/L-meq gene using nested set of primers for MDV-1
Virus Isolation
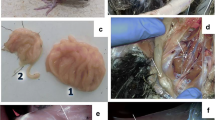
All nested PCR positive samples (n = 27) were processed for virus isolation; however, only seven (7) PCR positive samples showed formation of clear plaque, i.e., an area of cytopathic effect (CPE) with the aggregation of cells in CEF culture at 5th day post-infection (Fig. 3). These plaques were characteristics cytopathic effect (CPE) of Herpesvirus infection. Remaining samples did not show any CPE in cell culture. The TaqMan probe-based qPCR assay further confirmed the presence of the genome of the MDV-1 in all the harvested CPE producing cells as evident by Ct value of 28–34. Hence, the sensitivity of virus isolation in CEF was 25.9% (7/27). The details of the comparative result of qPCR, nPCR and virus isolation are given in Table 3.

Chicken embryo fibroblast culture after 5th day post-infection. A: Normal confluent monolayer cell CEF culture: MDV-1 Infected CEF with characteristic CPE
Nucleic Acid Sequencing and Sequence Analysis
A total of 14 nPCR amplicons including one long electropherotype were successfully sequenced and submitted to NCBI GenBank with accession numbers JN808273.1 to JN808279.1 and JN808281.1 to JN808287.1.Upon BlastN similarity search, the present nucleotide sequences of meq genes showed maximum similarity with meq gene sequences from different regions of the world. The nucleotide as well as deduced amino acid sequence identities of sequences in study along with other similar sequences from GenBank showed that present field strains belong to virulent and mild virulent strain of MDV-1 (Table 4). The neighbor-joining phylogenetic tree was constructed using deduced amino acid sequences of meq genes along with reference strain sequences from GenBank database (Fig. 4). The amino acid sequence alignment showed the variation in different strains of MDV-1 (Fig. 5).

Neighbor-joining phylogenetic tree showing relationship between Indian isolates of MDV-1with reference strain viruses

Multiple sequence alignment of meq gene amino acid sequences of MDV-1 strains
The molecular characterization of MDV-1 has been done by several authors, and it was proved that it can act as molecular marker for profiling of MDV-1 [28, 29]. In present study, percent identity of nucleotide as well as deduced amino acid sequences of Indian strains of MDV-1 and similar sequences from GenBank were calculated using Bioedit v7.2.5 program. Indian isolates ABT/HSR/487, ABT/HSR/5865, ABT/HSR/5924D, ABT/HSR/5924, ABT/HSR/5936, ABT/HSR/HSR477, ABT/HSR/HSR1512, ABT/HSR/2485, ABT/HSR/5936E, ABT/HSR/5937, ABT/HSR/8613 and ABT/HSR/13587 showed maximum nucleotide (nt) as well as amino acid (aa) identity of 99.6–100 (nt)/98.7–100(aa)% with reference virulent strain viruses from the USA (RB1B, GA and 571) [30] along with other virulent strains from India (MD/HYD/18/016,MD/HYD/18/018, MD/HYD/18/019, KeralaMty-MD-mx-1f, GADVASU-M3, LDH-3262), Turkey (MDV/Tur/2019) and Italy (GaHV-2/Italy/Ck/487/15) (Table 4). However, one of the Indian strains ABT/HSR/5129 showed 99.1–99.5/98–99.3% nt/aa identity with virulent strain viruses (Table 4). Thus, results revealed the high prevalence of virulent strains of MDV-1 in various districts of Haryana state. Moreover, one of the strain ABT/HSR/13181 showed maximum identity of 100/100 (nt/aa) with mild virulent reference strains from the USA (CU-2) and other USA strain (JM102) (Table 4). The strain ABT/HSR/13181 also showed maximum identity of 100/100 (nt/aa) with mild virulent vaccine strain viruses from China (814) and the Netherlands (CVI988) (Table 4). This indicated the close relationship of Indian ABT/HSR/13181 strain with vaccine viruses from the Netherland and China. In India, mostly bivalent live attenuated MD cell-associated vaccine is used which consists of ~ 5000 PFU of serotype 3 (HVT FC-126 strain) and ~ 3000 PFU of serotype 2(SB1 strain) per dose grown on chick embryo fibroblast cell culture. However, monovalent cell-free freeze-dried vaccine consisting of ~ 5000 PFU of serotype 3 (HVT FC-126 strain) is also in practice [11]. The live attenuated CVI988/Rispens strain of MDV-1 is used as gold standard vaccine globally [11]. It is much more efficient than recombinant vaccine. It has successfully proved its efficacy by decreasing the incidence of MD in poultry flock in the USA. However, it is not yet allowed for MDV-1 control among poultry flocks in India [11]. Therefore, it might be hypothesized that mild virulent strain (ABT/HSR/13181) had entered to Indian Territory somewhere in past either through vaccinated live birds or their products.
The phylogenetic analysis of deduced amino acid sequences revealed the four major clades, viz. virulent, very virulent, very virulent + and mild virulent (Fig. 4).The strain ABT/HSR/13181 (Accession number JN808274.1) was placed in mild virulent clad along with reference strains CVI988 from the Netherlands and 814 from China and CU-2 and JM102 from the USA. Remaining thirteen Indian strains were placed under virulent clad along with reference strains RB1B, GA and 571 from the USA and other strains from different regions of the world, viz. India (MD/HYD/18/016,MD/HYD/18/018, MD/HYD/18/019, KeralaMty-MD-mx-1f, GADVASU-M3, LDH-3262), Turkey (MDV/Tur/2019) and Italy (GaHV-2/Italy/Ck/487/15). Thus, percent sequence identity as well as phylogenetic analysis revealed the virulent strain as predominant strain and relatively low prevalence of mild virulent strain of MDV-1 in India. The PCR followed by nucleic acid sequencing also confirmed the outbreaks of virulent and very virulent strains in Tamil Nadu, Karnataka, Maharashtra and Andhra Pradesh state of India [15]. Similarly, the pathotyping of MDV-1 in live chicken birds also revealed the circulation of very virulent (Ind/TN/12/03) and virulent (Ind/TN/11/01, Ind/KA/12/02) strains in Southern Indian states [16].
Mutations in nucleotide sequence and corresponding change in amino acid sequence of meq gene lead to emergence of highly virulent oncogenic form of virus which is responsible for lymphoblastoma in different visceral organs. The amino acid sequence alignment of partial meq gene (142–286 aa) revealed the replacement of amino acid P with Q in virulent ABT/HSR/5129 strain (Fig. 5). There were no any changes detected in amino acid sequences among the mild virulent strains. However, there was an insertion of amino acid L was observed at position 146 in mild virulent strain (ABT/HSR/13181 and CU-2) in comparison to reference virulent strain GA [30] (Fig. 5). All the virulent strains showed 5 repeat regions of 4 prolines (PPPP) residue at amino acid position 152–155, 175–178, 191–194, 216–219 and 232–235 positions which is in coincidence with previous study[17]. 2018. However, in mild virulent strains, an additional PPPP repeats were observed at amino acid position 168–171 and that of at position 175–178 was lacking due to deletion of four amino acids PPPD at position 176–179. The number of repeats of four prolines residues (PPPP) in meq gene usually ranges in between two to eight which has already been correlated with oncogenicity and virulence in MDV-1. It is reported that most of the highly virulent strains of MDV-1 consist of lesser number of repeats and vice versa. Several researchers have identified 3–5 repeats in of PPPP residues in meq gene in Indian strains of MDV-1 [17]. Moreover, it has already been established that interruption in the PPPP repeats is associated with higher level of pathogenicity. Most virulent strains possess higher number of interruptions in PPPP repeats. In very virulent strain LMS [26] from China two of the PPPP repeats was interrupted at 175–178 (at position 176 P → R) and 216–219 (at position 217 P → A). Similarly, in very virulent + strain N from USA, three PPPP repeats were interrupted at 152–155 (at position 153 P → Q), 175–178 (at position 176 P → A) and 216–219 (at position 217 P → A). Previous study also revealed that interruption in repeats of proline residues at the second position increases the virulence and pathogenicity of MDV strains and highly virulent strains possess higher number of interruptions in PPPP residues at position 153, 176 and 276. The number of repeats in PPPP residues was also reflected during phylogenetic study. Virulent strains and mild virulent strains possessed five repeats at different positions and formed separate individual clads. Similarly, very virulent and very virulent + strains possessed three and two repeats, respectively, and formed separate individual clads (Fig. 4).
In spite of intensive vaccination, still severe outbreaks of MDV-1 leading to huge economic losses are reported from India [12, 17]. Therefore, molecular characterization of MDV-1 is essential for monitoring the changes in genome of viruses and evaluating the effectiveness of existing vaccines. Recently, monovalent (HVT) or bivalent (SB-1 and HVT) strains are used as vaccine for the control of virulent strains of MDV-1. Moreover, there is a paradigm shift in clinical manifestation of diseases from classical or paralytic form to an acute lymphomatosis in the affected birds is also reported which might be attributed to emergence of highly virulent MDV-1 strains.
Conclusion
The present study demonstrated real-time PCR as most sensitive molecular assay as compared to nPCR and virus isolation for the detection of MDV-1 in infected liver sample of poultry. The meq gene-based nested PCR and real-time PCR can be used for monitoring of MDV-1 outbreak. Nucleotide sequence analysis of meq gene revealed the circulation of virulent MDV-1strain among poultry population in Haryana. Thus, in context of emergence of more virulent form of MDV-1, it is recommended for use of more efficient and updated MDV-1 vaccine developed from surveillance data of Indian MDV-1 strains in the poultry farms for prevention and control of the disease.
References
Bulow VV, Biggs PM (1975) Differentiation between strains of Marek’s disease virus and turkey herpes virus by immunofluorescence assays. Avian Pathol 4:133–146
Witter RL, Calnek BW, Buscaglia G, Gimeno IM, Schat KA (2005) Classification of Marek’s disease viruses according to pathotype: Phylosophy and Methodology. Avian Pathol 34:75–90
Sung HW (2002) Recent increase of Marek’s disease in Korea related to the virulence increase of the virus. Avian Dis 46:517–524
Atkins KE, Read AF, Savill NJ, Renz KG, Fakhrul Islam AFM et al (2013) Vaccination and reduced cohort duration can drive virulence evolution:Marek’s disease virus and industrialized agriculture. Evolution 67(3):851–860
Calnek BW (2001) Pathogenesis of Marek’s disease virus infection. In: Hira K (ed) Current topics in microbiology and immunology. Springer, Berlin, pp 25–56
Nair V (2005) Evolution of Marek’s disease- A paradigm for incessant race between the pathogen and the host. Vet J 170:175–183
Couteaudier M, Denesvre C (2014) Marek’s disease virus and skin interactions. Vet Res 45(1):36
Jones D, Lee L, Liu JL, Kung HJ, Tillotson JK (1992) Marek disease virus encodes a basic-leucine zipper gene resembling the fos/jun oncogenes that is highly expressed in lymphoblastoid tumors. Proc Natl Acad Sci USA 89:4042–4046
Liu JL, Kung HJ (2000) Marek’s disease herpesvirus transforming protein MEQ: a c-Jun analogue with an alternative life style. Virus Genes 21:51–64
Renz KG, Cooke J, Clarke N, Cheetham BF, Hussain Z, Fakhrul Islam AF, Tannock GA, Walkden-Brown SW (2012) Pathotyping of Australian isolates of Marek’s disease virus and association of pathogenicity with meq gene polymorphism. Avian Pathol 41:161–176
Kannaki TR, Vasudevan G Marek’s disease: time to review the emerging threat in Indian poultry. World's Poultry Sci J. https://doi.org/10.1080/00439339.2020.1729674
Muniyellappa HK, Satyanarayana ML, Isloor S, Shivakumar Gowda NK (2013) Marek’s disease outbreak among vaccinated commercial layer flocks in the mining area of Karnataka, India. Vet Rec 172(17):452. https://doi.org/10.1136/vr.101203
Bhutia LD, Damodar SY (2017) Marek’s disease outbreak in poultry population of Mizoram. India Indian J Vet Patholo 41(3):224–227
Kamaldeep, Sharma PC, Narang G (2007) Occurrence of Marek`s disease in vaccinated poultry flocks of Haryana (India). Int J Poultry Sci 6(5): 372–377
Raja A, Dhinakar Raj G, Bhuvaneswari P, Balachandran C, Kumanan K (2009) Detection of virulent Marek’s disease virus in poultry in India. Acta Virol 53(4):255–260
Suresh P, Johnson Rajeswar J, Sukumar K, Harikrishnan TJ, Srinivasan P (2015) Pathotyping of recent Indian field isolates of Marek’s disease virus serotype 1. Acta Virol 59(2):156–165
Puro K, Bhattacharjee U, Baruah S, Sen A, Das S, Ghatak S, Shakuntala I (2018) Characterisation of Marek’s disease virus and phylogenetic analyses of meq gene from an outbreak in poultry in meghalaya of Northeast India. Virus Disease. 29:167–172
Baigent SJ, Smith LP, Currie RJ, Nair V (2005) Replication kinetics of Marek’s disease vaccine virus in feathers and lymphoid tissues using PCR and virus isolation. J Gen Virol 86:2989–2998
Murata S, Hayashi Y, Kato A, Isezaki M, Takasaki S, Onuma M, Osa Y, Asakawa M, Konnai S, Ohashi K. Surveillance of Marek's disease virus in migratory and sedentary birds in Hokkaido, Japan. 2012; Vet J. 192(3):538–540.
Hall TA BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 1999(35):1547–1549
Baigent SJ, Nair VK, Le Galludec H (2016) Real-time PCR for differential quantification of CVI988 vaccine virus and virulent strains of Marek’s disease virus. J virol methods 233:23–36. https://doi.org/10.1016/j.jviromet
Abdul-Careem MF, Hunter BD, Nagy E, Read LR, Sanei B, Spencer JL, Sharif S (2006) Development of a real-time PCR assay using SYBR Green chemistry for monitoring Marek’s disease virus genome load in feather tips. J Virol Methods 133(1):34–40
Islam A, Harrison B, Cheetham BF, Mahony TJ, Young PL, Walkden-Brown SW (2004) Differential amplification and quantitation of Marek’s disease viruses using real-time polymerase chain reaction. J Virol Methods 119(2):103–113
Davidson I, Natour-Altoury A, Raibstein I, Dahan Y (2017) Differential amplification of Marek’s disease CVI988 vaccine and of wild-type isolates from organs of commercial chickens using single or duplexed probes in real-time PCR. Avian Pathol 46(6):610–614
Cheng Y, Cong F, Zhang YP, Li ZJ, Xu NN, Hou GY, Liu CJ (2012) Genome sequence determination and analysis of a Chinese virulent strain, LMS, of Gallid herpesvirus type 2. Virus Genes 45(1):56–62
Schat KA (2005) Isolation of Marek’s disease virus: revisited. Avian Pathol 34(2):91–95
Mohamed MH, El-Sabagh IM, Al-Habeeb MA, Al-Hammady YM (2016) Diversity of Meq gene from clinical Marek’s disease virus infection in Saudi Arabia. Vet World 9(6):572–578
López-Osorio S, Piedrahita D, Espinal-Restrepo MA, Ramírez-Nieto GC, Nair V, Williams SM, Baigent S, Ventura-Polite C, Aranzazu-Taborda DA, Chaparro-Gutiérrez JJ (2017) Molecular characterization of Marek’s disease virus in a poultry layer farm from Colombia. Poult Sci 96(6):1598–1608. https://doi.org/10.3382/ps/pew464
Coussens PM, Velicer LF (1988) Structure and complete nucleotide sequence of the Marek’s disease herpesvirus gp57-65 gene. J Virol 62(7):2373–2379
Acknowledgements
The poultry farmers, field veterinarians and non-teaching staff of the Department of Animal Biotechnology are acknowledged for their help in the study. We acknowledge funding support from LUVAS Hisar.
Funding
LUVAS, State Scheme Number 121.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Significance statement: Marek’s disease (MD) is a lymphoproliferative and neuropathic disease of domestic fowl caused by an oncogenic Gallid Herpesvirus-2 (GaHV-2), also known as Marek’s Disease Virus serotype 1 (MDV-1). Present study has explained the circulation of virulent strain of MDV-1, responsible for morbidity and mortality among vaccinated poultry birds in Haryana state of India. It has not been published elsewhere and that it has not been submitted simultaneously for publication elsewhere.
Rights and permissions
About this article

Cite this article
Kumar, A., Maan, N.S., Mahajan, N.K. et al. Isolation and Molecular Characterization of MDV-1 from Vaccinated Poultry Birds in Haryana State of India. Proc. Natl. Acad. Sci., India, Sect. B Biol. Sci. 92, 679–690 (2022). https://doi.org/10.1007/s40011-022-01363-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40011-022-01363-1