
Abstract
Purpose
This study investigated the metabolism and pharmacokinetics of apigenin and its metabolite, apigenin-7-O-glucuronide (A7G), using a newly developed UPLC-MS/MS method.
Methods
A simple and sensitive UPLC-MS/MS method was developed for simultaneous quantification of apigenin and A7G, and their pharmacokinetic properties were examined using an in vivo rat model. Moreover, stability under various conditions, protein binding, blood distribution, and metabolic studies were performed in vitro.
Results
Apigenin showed poor stability in simulated intestinal fluid, whereas A7G was stable for 24 h. An in vivo pharmacokinetic study demonstrated that apigenin had a very low oral bioavailability (F) of 0.708% and was mainly metabolized to A7G. Notably, systemic exposure (Cmax and AUC) of apigenin after oral administration of A7G was markedly higher (2.62- and 14.3-fold, respectively) than that after oral administration of apigenin. Apigenin and A7G were significantly metabolized in both hepatic and intestinal S9 fractions. Based on the well-stirred and QGut model concepts, they were classified as compounds with low EH (0.0167–0.0389) and moderate-to-high EG (0.626–0.979) in rats, indicating that the intestine had a greater contribution than the liver to pre-systemic elimination of both phytochemicals.
Conclusion
The low F of apigenin could be attributed to its poor stability in the gastrointestinal lumen and extensive intestinal first-pass effect, which could be improved by oral administration of A7G, demonstrating the potential of A7G as a natural prodrug for improving the low F of apigenin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, natural products and their potential health benefits have garnered substantial attention from the scientific community (Gorzynik-Debicka et al. 2018; Chang et al. 2019; Vucic et al. 2019). Among these natural products, flavonoids have emerged as a class of compounds of particular interest owing to their diverse pharmacological properties and potential therapeutic applications (Fraga et al. 2019; Cui et al. 2022; Rana et al. 2022). Apigenin is a well-known flavonoid with various beneficial effects on human health and exhibits anticancer, anti-inflammatory, antioxidant, antidepressant, antimutagenic, antiviral, and hepatoprotective properties (Serafini et al. 2010; Prochazkova et al. 2011; Kumar et al. 2013; Gontijo et al. 2017; Kashyap et al. 2022). Compared to structurally similar flavonoids, apigenin has lower intrinsic toxicity to normal cells than cancer cells and can suppress tumor development by inhibiting the proliferation of cancer cells (Salehi et al. 2019). Apigenin exerts anticancer activity through various mechanisms, including activating proteasome degradation of Her2/neu protein, inactivating NF-κB, and regulating various kinase activities (Imran et al. 2020). Apigenin-7-glucuronide (A7G) is derived from apigenin and is present in many fruits and vegetables, including celery, parsley, artichoke, and cardoon, some of which are widely sold as dietary and herbal supplements (Marrassini et al. 2020). A7G has attracted substantial interest because of its promising biological activities, including anti-inflammatory, antioxidant, antidiabetic, antiviral, and anti-Alzheimer properties (Cheng et al. 2013; Jin et al. 2022; Kurnia et al. 2023). Notably, it inhibits the overexpression of matrix metalloproteinases implicated in the development of cancer, arthritis, Alzheimer’s disease, and cardiovascular diseases (Crasci et al. 2017).
Based on its physicochemical and pharmacokinetic properties, apigenin is classified as a BCS class 2 drug owing to its low solubility and high intestinal permeability (Liu et al. 2002; Zhang et al. 2012). Apigenin is absorbed through the entire intestine and primarily through the duodenum. Following oral administration of apigenin in humans, less than 0.5% of the dose is excreted as metabolites via urine over 24 h (Borges et al. 2022). Apigenin is metabolized by cytochrome P450-mediated phase I reactions (to form luteolin, scutellarein, and isoscutellarein) and/or phase II reactions such as glucuronide and sulfate conjugation (Gradolatto et al. 2004). A portion of conjugated apigenin can be excreted via bile, hydrolyzed by intestinal β-glucuronidase, and then reabsorbed as aglycone (Wang et al. 2019; Gao et al. 2022). Flavonoids have numerous health benefits; however, their utility as bioactive substances is limited in the clinical setting because of their low oral bioavailability (F) (Seo et al. 2022a). F value is influenced by factors such as solubility, stability in the gastrointestinal tract, intestinal absorption, and intestinal/hepatic first-pass effect. Therefore, it is important to identify the mechanism underlying low F value to develop effective oral formulations. Although a few studies have reported the pharmacokinetic properties of apigenin (Gradolatto et al. 2005; Wan et al. 2007), little information is currently available on the quantitative mechanism underlying the low F of apigenin. Moreover, to the best of our knowledge, pharmacokinetic studies on A7G, the major active metabolite of apigenin, are scarce.
Several studies have described bioanalytical methods for apigenin using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) and high-performance liquid chromatography coupled with UV/Vis detector (Li et al. 2005; Wan et al. 2007; Elzayat et al. 2019; Zhu et al. 2021). In addition, the concentration of A7G in biological samples has been analyzed using LC-MS/MS (Tu et al. 2020; Wang et al. 2023). Additionally, several studies have attempted to quantify the total flavonoid content (including aglycones and their conjugates) by pretreating biological samples via hydrolysis reaction (Shia et al. 2010; Trontelj. 2012; Han et al. 2022). However, this method may be inaccurate because of the physicochemical instability of flavonoids during hydrolysis reaction; moreover it does not offer simultaneous quantification of aglycone and its conjugates (Ding et al. 2013). To date, no bioanalytical methods for simultaneously determining apigenin and its metabolites have been reported, warranting further study.
In this study, a simple and sensitive method was developed for simultaneous quantitative analysis of apigenin and A7G in rat plasma using ultra-performance liquid chromatography coupled with a tandem mass spectrometry (UPLC-MS/MS) system. The performance and reproducibility of this bioanalytical method have been fully validated according to the FDA guidelines (title: Bioanalytical Method Validation; website: https://www.fda.gov/media/70858/download). Next, an in vivo pharmacokinetic study of apigenin and A7G was performed using rat model and validated method. Additionally, factors affecting the F of apigenin and A7G, such as physicochemical stability and hepatic/intestinal metabolic clearance, were investigated using the well-stirred and Qgut models.
Materials and methods
Materials and animals
Apigenin (> 97% purity) and A7G (> 98% purity) were purchased from ChemFaces (Hubei, China). Alpelisib (> 99% purity; internal standard (IS)) was obtained from MedKoo Bioscience, Inc. (Morrisville, NC, USA). Adenosine 3’-phosphate 5’-phosphosulfate lithium salt hydrate (PAPS), carboxymethylcellulose, dimethyl sulfoxide (DMSO), formic acid (FA), triphosphopyridine nucleotide reduced tetrasodium salt (NADPH), S-adenosyl methionine (SAM), and uridine-diphosphate-glucuronic acid trisodium salt (UDPGA) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Alamethicin was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). Pooled rat hepatic and intestinal S9 fractions were obtained from Sekisui Xenotech (Kansas City, KS, USA). Adult male Sprague-Dawley (SD) rats (mean body weight: 264 g) were supplied by DAEHAN BIOLINK Co., Ltd. (Chungcheong Buk-do, Korea). All animals were used following an adaptation period of one week, and diet except water was restricted from the afternoon of the day before the experiment. This study was performed according to the protocol approved by Institutional Animal Care and Use Committee of Pusan National University (approval number: PNU-2023-3245; Busan, Republic of Korea).
Analytical equipment
The bioanalysis method was validated using an UPLC-MS/MS system comprising two LC-30AD pumps, SIL-30AC autosampler, CTO-20AC column oven, and LCMS-8050 mass detector. Separation of apigenin, A7G, and IS was achieved on Kinetex® column (100 × 2.1 mm, 2.6 μm, 100 Å) and SecurityGuard Cartridge C18 pre-column (SecurityGuard HPLC cartridge system). The mobile phase gradient was as follows: [solvent A: deionized water (0.1% FA); solvent B: acetonitrile (0.1% FA)]: 0.01–6.00 min 15.0–40.0% B, 6.01–8.00 min, 40.0–95.0% B, 8.00–8.01 min 95.0–40.0% B, 8.01–10.0 min, 15% B (flow rate of mobile phase was 0.3 mL/min; total analysis time was 10 min). The column oven was set as 40 °C. The following ion source parameters were set: desolvation temperature, 250 °C; drying gas flow, 10 L/min; heating block temperature, 400 °C; heating gas flow, 10 L/min; interface temperature, 300 °C; and nebulizing gas flow, 3 L/min.
Calibration standard and quality control sample preparation
Stock solutions of apigenin and A7G (1 mg/mL) were prepared with methanol and stored at − 20 °C. The stock solutions were serially diluted using methanol to prepare working standard solutions of concentrations ranging from 0.2 to 200 µg/mL for apigenin and A7G. The final concentrations for each calibration standard sample were 2, 5, 10, 20, 50, 100, 200, 500, 1000, and 2000 ng/mL for apigenin and A7G. Quality control (QC) samples were prepared from separate sets of stock solutions at 2 ng/mL (low limit of quantification, LLOQ), 6 ng/mL (low QC; LQC), 150 ng/mL (medium QC, MQC), and 1500 ng/mL (high QC, HQC) for apigenin and A7G. Alpelisib (IS) was prepared at a concentration of 1 mg/mL in methanol, and the final IS spiking solution contained 10 ng/mL IS in acetonitrile. Aliquots of equal volumes of 5% FA were added to 50 µL of samples in a 1.5 mL micro-centrifuge tube. Each mixture was then vortexed for 1 min. Then, an aliquot of 300 µL of acetonitrile containing 10 ng/mL of IS was deproteinized by adding the mixtures (Yun et al. 2023), vortexed for 1 min, and centrifuged at 12,500 × g for 10 min at 4 °C. The supernatant (300 µL) was collected and evaporated using SpeedVac (Eyela). The residue was re-dissolved in 50 µL of methanol. After centrifuging at 12,500 × g for 5 min, 3 µL of supernatant was injected into the UPLC-MS/MS system fot quantification of apigenin and A7G.
Method validation
According to the US FDA guidelines, the bioanalytical method for apigenin and A7G has been fully validated for selectivity, linearity, accuracy, precision, recovery, matrix effect, dilution integrity, and stability (US Food and Drug Administration. 2018). The selectivity for apigenin, A7G, and IS was assessed using blank plasma from three animals and evaluated by comparing the chromatograms of blank plasma spiked with IS alone, blank plasma spiked with apigenin and IS, blank plasma spiked with A7G and IS, and blank plasma spiked with apigenin, A7G, and IS. The linearity of analysis was evaluated for a concentration range of 2–2000 ng/mL, using standard samples prepared from plasma for both apigenin and A7G. The calibration curves for apigenin and A7G were prepared using the peak area ratios of apigenin and IS or A7G and IS by weighted (1/x) linear regression analysis, and the r2 value of the calibration curve was used to assess linearity. Accuracy and precision were evaluated using QC samples, including LLOQ-spiked rat plasma, on five different days. According to the US FDA guideline acceptance criteria, accuracy and precision were set within ± 15% of coefficient variation (CV), respectively, except at the LLOQ, which were set within ± 20%. Extraction recovery was evaluated to determine whether the extraction of apigenin, A7G, and IS from plasma by deproteinization was efficient and reproducible. The peak areas of apigenin and A7G extracted from plasma samples (sample A) spiked with apigenin and A7G before extraction were compared with those of samples spiked with apigenin and A7G after deproteinization (sample B). The matrix effect was evaluated using LQC, MQC, and HQC to determine whether endogenous plasma components affected the ionization of apigenin, A7G, and IS. To determine the matrix effects of apigenin and A7G, the average peak area of sample B was compared with that of neat acetonitrile solution (sample C). The extraction recovery was calculated as ‘A/B × 100’, and the matrix effect was calculated as ‘B/C × 100’. Five replicates were assessed at each QC level. The dilution integrity of apigenin and A7G was evaluated five times after 100-fold diluting plasma control sample at a very high concentration of 200000 ng/mL. The analytical stability of apigenin and A7G in rat plasma was assayed by evaluating the autosampler (post-preparative stability), benchtop, freeze-thaw, and long-term stabilities at LQC and HQC in five replicates. The autosampler stability was determined by exposing pretreated plasma samples at room temperature (20–23 °C) for 24 h in the autosampler. Benchtop stability was determined after exposing the spiked plasma samples to room temperature for 3 h. Freeze-thaw stability was determined under three freeze-thaw cycles for three days. In addition, long-term stability was assessed by measuring spiked plasma samples stored at − 70 °C for 4 weeks.
In vitro protein binding, blood distribution, and physicochemical stability studies
In vitro protein binding (plasma, rat hepatic S9 fraction, and intestinal S9 fraction) and the plasma concentration ratio (RB) were determined as previously described (Seo et al. 2022a, 2022b). For apigenin, the protein concentration in the hepatic and intestinal S9 fractions used for protein binding was 0.2 mg/mL, whereas for A7G, the protein concentration in the hepatic and intestinal S9 fractions used for protein binding was 1 mg/mL. The physicochemical stability of apigenin and A7G was determined using various pH buffers (pH 1.0–11.0) and biological samples including plasma, urine, simulated gastric fluid (SGF), and simulated intestinal fluid (SIF). SGF (final pH 1.2) was prepared using 0.7% hydrochloride, 0.2% sodium chloride, and 0.32% pepsin. SIF (final pH 6.8) was prepared using 3 mM sodium taurocholate in a phosphate buffer containing 0.1% pancreatin. After incubating for 0, 15, 30, 60, 120, 240, 480, and 1440 min in a glass vial at 37 °C, 50 µL of samples were placed in the prepared 1.5 mL micro-centrifuge tube containing IS, and the samples were analyzed using UPLC-MS/MS.
In vivo pharmacokinetic study in rats
The developed UPLC-MS/MS method was successfully used to investigate the pharmacokinetics of apigenin and A7G in rats. Cannulas were surgically implanted into the femoral vein and artery of rats under anesthesia, as described previously (Seong et al. 2022; Vo et al. 2022; Keem et al. 2023). Apigenin and A7G were dissolved in DMSO, ethanol, polyethylene glycol 400, and 0.5% carboxymethylcellulose solution (1:5:80:14, v/v/v/v) for intravenous and oral dose. Apigenin was administered intravenously at a single dose of 20 mg/kg and orally at a single dose of 30 mg/kg, or A7G was administered intravenously at a single dose of 33.1 mg/kg (20 mg/kg as apigenin) and orally at a single dose of 49.6 mg/kg (30 mg/kg as apigenin). Blood samples were collected from the femoral artery at 0, 2, 5, 15, 30, 60, 90, 120, 240, and 480 min after intravenous administration and at 0, 2, 5, 15, 30, 60, 90, 120, 180, 240, 360, and 480 min after oral administration. The total volume of blood collected during the intravenous and oral pharmacokinetic studies was 2.1 mL, which was less than 10% of the total circulating blood volume in rats. The blood samples were immediately placed in a centrifuge tube, and the plasma was separated by centrifugation at 9000 × g for 2 min at 4 °C. The obtained plasma samples were transferred to a new 1.5 mL micro-centrifuge tube and stored at − 70 °C until UPLC-MS/MS analysis. Urine samples were collected in light-protected 15 mL tubes for up to 24 h after administration. The entire gastrointestinal content (including feces) samples were obtained as previously described (Seo et al. 2023). Briefly, rats were sacrificed, and the gastrointestinal tract was removed via laparotomy. The extracted gastrointestinal tract was placed in a beaker containing methanol and cut into small pieces using scissors to extract analytes.
In vitro metabolism study in rat S9 fraction
The metabolism of apigenin and A7G was determined as previously described (Seo et al. 2023). Briefly, composition of the culture mixture was as follows: substrate (apigenin or A7G), rat hepatic/intestinal S9 fractions (protein concentration: 0.2 mg/mL for apigenin, 1 mg/mL for A7G), 10 mM MgCl2, and cofactor (1 mM NADPH, 2 mM UDPGA, 5 mM SAM, 0.04 mM PAPS) in 0.1 M Tris buffer (pH 7.4). The S9 fraction was used here because it is routinely used in the metabolism studies, particularly for phase II metabolic pathways involving sulfation (Tang et al. 2012). The reaction was initiated by the addition of cofactors after pre-incubation for 5 min at 37 °C. For the control group, equivalent volumes of water were added instead of cofactors. After incubation at 37 °C for 0, 15, 30, 60, and 120 min, the reaction was terminated by adding 50 µL of 5% FA and 300 µL of ice-cold acetonitrile containing IS. Following the preparation described above, the samples were analyzed using UPLC-MS/MS. Hepatic and intestinal CLint,S9 were calculated using the half-life (t1/2) method (Seo et al. 2022a, 2022b, 2023) for metabolic stability test. The t1/2 of apigenin and A7G in the S9 fraction was estimated from the slope of the incubation time against the log residual percentage profile, and the CLint,S9 of apigenin and A7G was estimated using the following equation (Seo et al. 2022a):
where fuS9 is the unbound fraction of apigenin or A7G in the hepatic and intestinal S9 fractions, respectively. Hepatic and intestinal CLint,S9 estimated in vitro was scaled to the organ level using the scaling factors reported in the literature.
Pharmacokinetic analysis
The pharmacokinetics of apigenin and A7G in rats were evaluated by a non-compartmental analysis using WinNonlin software (ver. 3.1, NCA 200 and 201). The following pharmacokinetic parameters were assessed: total area under the plasma concentration versus time curve from time zero to infinity (AUC) and from time zero to the last measurable time (AUClast), terminal half-life (t1/2), apparent volume of distribution at steady state (Vss), and total body clearance (CL). The peak plasma concentration (Cmax) and the time to reach Cmax (Tmax) were obtained directly from the plasma concentration versus time data. The dose fraction of the drug converted into a certain metabolite (Fm) was estimated as follows (Iwaki et al. 1989):
where, AUC’x is the AUC of metabolite following the administration of the parent drug, and AUC’ is the AUC following intravenous administration of an equimolar dose of the metabolite. F is the proportion of the dose absorbed after oral administration (Fabs), intestinal availability (FG), and hepatic availability (FH) as shown in the following equation (Yang et al. 2007):
EH can be predicted using the following well-stirred model (Yoon et al. 2011):
The QGut model can be used to predict first-pass EG from intestinal permeability and clearance data. The QGut model was incorporated to describe the gut first-pass extraction process as follows (Yang et al. 2007):
where, Qvilli and CLperm represents the mucosal blood flow and permeability clearance, respectively. CLperm was calculated by multiplying the intestinal surface area (Aintestine) with the effective permeability of the intestine (Peff) (Yoon et al. 2011). Fabs was estimated by comparing Aegi values obtained from in vivo pharmacokinetic analysis, as shown in the following equation (Seo et al. 2022a, 2022b):
Results
Method development
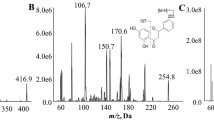
To quantify plasma apigenin and A7G, the full mass spectrum was scanned for positive and negative ionization, with better sensitivity being observed for positive ions. Precursor [M + H]+ ion signals were observed at m/z 271.05 for apigenin, m/z 447.10 for A7G, and m/z 442.10 for IS. These precursor ions were further fragmented, and the most abundant product ions were observed at m/z 153.05 for apigenin, m/z 271.10 for A7G, and m/z 328.00 for IS (Fig. 1). The following optimized LC conditions were used: Kinetex C18 column was used as the stationary phase, and gradient elution program was used for mobile phase system consisting of water, ACN, and FA. The retention time under optimized LC conditions was 5.11 min for apigenin, 2.87 min for A7G, and 5.78 min for IS. As shown in Fig. 2, all analyte peaks had acceptable shapes and resolutions. Plasma samples were pretreated with acetonitrile using a simple protein precipitation method. FA was added to improve the recovery rate and efficiency of the pre-treatment method for biological samples. Thus, the method was optimized by adding 50 µL of FA and 300 µL of acetonitrile containing IS to 50 µL of plasma.

Product ion scan mass spectra of a apigenin, b A7G, and c IS

Representative LC-MS/MS chromatograms of apigenin, A7G, and IS in rat plasma samples: a blank rat plasma; b blank rat plasma spiked with analytes at the LLOQ (2 ng/mL); c blank rat plasma spiked with analytes at the MQC (150 ng/mL); d plasma sample collected 15 min after oral administration of apigenin at a dose of 30 mg/kg in rats
Method validation
Calibration curves for apigenin and A7G were obtained by plotting the apigenin peak area/IS peak area versus concentration and the A7G peak area/IS peak area versus concentration, respectively. Apigenin and A7G showed excellent linearity at concentrations of 2–2000 ng/mL (r2 > 0.999). The accuracy for apigenin was 81.9–118%, and that for A7G was 82.2–118%, with a precision of < 14.4% for apigenin and < 11.9% for A7G (Table 1). The developed assay accurately and reproducibly quantified apigenin and A7G in rat plasma. Extraction recoveries for apigenin and A7G from rat plasma were 89.7–107% (CV < 6.02%) and 90.7–111% (CV < 10.8%), and the matrix effects were 95.3–111% (CV < 5.75%) and 88.4–113% (CV < 7.18%), respectively. The values for extraction recovery and matrix effects were within acceptable ranges, indicating consistent and excellent extraction recovery with no matrix effect in rat plasma. With regard to the dilution integrity of apigenin and A7G, the precision/accuracy of plasma control samples at 200000 ng/mL (100 times of the ULOQ) were 8.61/101 ± 9% for apigenin and 12.5/103 ± 13% for A7G, following 100-fold dilution. The stability values of the plasma samples are listed in Table 2. This indicates that apigenin and A7G remained stable under all controllable conditions. Therefore, the developed method is accurate and reproducible under all feasible conditions.
In vitro protein binding, blood distribution, and physicochemical stability studies
The unbound fractions of apigenin and A7G in plasma (fuP) were 0.0101 ± 0.0017 and 0.108 ± 0.008, respectively, indicating that apigenin exhibits extensive protein binding compared with that of A7G. The unbound fractions of apigenin and A7G in the rat hepatic and intestinal S9 fractions (fuHS9 and fuIS9, respectively) were 0.801 ± 0.033 and 0.796 ± 0.039, and 0.895 ± 0.023 and 0.898 ± 0.016, respectively. These results indicated that both compounds exhibited low protein binding. The RB values of apigenin and A7G were 2.80 ± 0.11 and 2.45 ± 0.11, respectively, indicating that they are mainly distributed in the plasma rather than in red blood cells. The stability profiles of apigenin and A7G under various conditions are shown in Fig. 3. Apigenin was relatively stable for 24 h at pH 1–7, with 86.0–104% remaining, but was slightly unstable at pH 9 and 11 with only 67.2–82.1% remaining after 24 h. Additionally, in biological samples, apigenin was slightly unstable in plasma, urine, and SGF, with 43.0–71.4% remaining after 24 h; however, it was highly unstable in SIF, with only 0.933–1.42% remaining after 24 h. In contrast, A7G was stable for 24 h in various acidic and basic pH buffers and was stable for 24 h in plasma, urine, SGF, and SIF.

Stability of apigenin in a buffers of different pH values (pH 1, 3, 5, 7, 9, and 11) and b biological samples (plasma, urine, SGF, and SIF). Stability of A7G in c buffers of different pH values (pH 1, 3, 5, 7, 9, and 11) and d biological samples (plasma, urine, SGF, and SIF) (n = 3)
In vivo pharmacokinetic study in rats
The plasma concentration-time profiles following intravenous and oral administration of apigenin and of A7G in rats are shown in Fig. 4, and the relevant parameters are listed in Tables 3 and 4. Following intravenous administration of apigenin, plasma apigenin concentration decreased in a multi-exponential manner, with Vss of 643–1197 mL/kg showing low-to-moderate tissue distribution. Apigenin was rapidly metabolized to A7G following intravenous administration, with Cmax of A7G as high as 32414–54290 ng/mL. Overall, 4.65–9.24% of apigenin dose was excreted in urine and 1.05–7.83% in feces. Apigenin was rapidly absorbed after oral administration, with Tmax of 5 min and Cmax of 113–309 ng/mL, followed by a multi-exponential decrease to a t1/2 of 114–167 min. The F was estimated to be 0.00708. Following oral administration of apigenin, the Tmax and Cmax of A7G were 5–15 min and 521–697 ng/mL, respectively, indicating that apigenin was rapidly metabolized to A7G in rats. Overall, 0.095–0.393% of apigenin dose was excreted in urine and 23.3–59.0% in feces. Following intravenous administration of A7G, low concentrations of apigenin were detected in the plasma for up to 30–60 min, with a t1/2 of 6.72–24.4 min. A7G decreased in a multi-exponential manner with a t1/2 of 134–255 min, and Vss and CL were 994–1812 mL/kg and 20.1–26.5 mL/min/kg, respectively. Overall, 0.368–4.56% of the A7G dose was excreted in urine and 0.0742–3.60% of in feces. Tmax of apigenin after oral administration of A7G was 180–240 min, which was slower than that after oral administration of apigenin, however, Cmax was 2.62 times higher, although there was no significant difference between the two groups. In addition, the AUC of apigenin after oral administration of A7G was 82.0 ± 43.4 µg·min/mL and was significantly higher (14.3 fold) than that after oral administration of apigenin. However, because the terminal phase of A7G could not be determined after oral administration of A7G, the exact F of A7G could not be determined. Therefore, the F of A7G was determined to be 0.121 based on AUClast. Overall, 0.536–5.44% of A7G dose was excreted in urine and 4.74–15.2% in feces.

Mean plasma concentration-time profiles of apigenin and A7G in rats following a intravenous administration of apigenin; b oral administration of apigenin; c intravenous administration of A7G; and d oral administration of A7G (n = 4–5)
In vitro metabolism study in rat S9 fraction
The metabolism of apigenin has been described in a previous study; however, the metabolism of A7G has not yet been elucidated (DeRango-Adem et al. 2021). Therefore, to evaluate the metabolic contribution and kinetics of apigenin and A7G in the liver and intestine, phase I (NADPH) and phase II (glucuronidation, methylation, and sulfation) metabolism were investigated using hepatic and intestinal S9 fractions. As shown in Fig. 5, the logarithmic fractions of residual apigenin and A7G versus time curves declined mono-exponentially, indicating that the metabolism of apigenin and A7G followed first-order (linear) kinetics. The residual apigenin was 1.10–1.24% in the hepatic S9 fraction after 120 min in the presence of cofactor, and 81.4–86.9% in the intestinal S9 fraction after 120 min in the presence of cofactor. In the hepatic S9 fraction, t1/2 was estimated as 28.6 ± 0.7 min, and hepatic CLint,S9 was estimated as 0.151 ± 0.004 mL/min/mg protein. In the intestinal S9 fraction, t1/2 was estimated as 537 ± 66 min, and intestinal CLint,S9 was estimated as 0.00817 ± 0.00107 mL/min/mg protein. These results indicate that apigenin is metabolized in the liver and intestine. In contrast, there was no significant difference in A7G, regardless of the presence or absence of cofactor, and residual A7G was 33.9–44.1% in the hepatic S9 fraction, regardless of the presence or absence of the cofactor. Furthermore, residual A7G was 82.4–91.7% in the intestinal S9 fraction, indicating that NADPH-mediated phase I metabolism, glucuronidation, methylation, and sulfation were not involved in the metabolism of A7G. Considering its stability in the buffers tested, it is assumed that A7G was degraded through enzymatic hydrolysis in the liver and intestine. In the hepatic S9 fraction, t1/2 was estimated as 82.6 ± 9.8 min, and hepatic CLint,S9 was estimated as 0.00947 ± 0.00101 mL/min/mg protein. In the intestinal S9 fraction, t1/2 was estimated as 505 ± 102 min, and intestinal CLint,S9 was estimated as 0.00157 ± 0.00026 mL/min/mg protein. The hepatic and intestinal CLint,S9 of apigenin and A7G estimated in vitro were scaled up to the CLint values for the whole organs (Table 5).

Time course of remaining fraction of apigenin (a) and A7G (b) in rat hepatic and intestinal S9 fractions, respectively (n = 5). The bullet symbol represents the mean, the error bar represents the standard deviation, and the asterisk indicates value significantly different from that of the control (i.e., the remaining fractions measured at 0 min) (P < 0.05)
Discussion
Our study aimed to develop a simple and sensitive UPLC-MS/MS method to quantify apigenin and its major active metabolite, A7G, in rat plasma and investigate the factors determining the F of apigenin and A7G. We developed an efficient and sensitive bioanalytical method for simultaneous quantification of apigenin and A7G, meeting the US FDA criteria for linearity, selectivity, accuracy, precision, recovery, matrix effect, dilution integrity, and stability. This showed that the developed method was accurate, precise, and reproducible, with high recovery and minimal matrix effects. Several bioanalytical methods for analyzing apigenin or A7G in rat plasma have been reported (Tu et al. 2020; Zhu et al. 2021; Wang et al. 2023); however, no methods have been reported for the simultaneous determination of these two phytochemicals. Compared to existing studies, our study findings suggest an efficient, time-saving, and cost-effective method for simultaneous analysis of parent drug and primary metabolite.
The intravenous and oral doses of apigenin for in vivo pharmacokinetic study were selected based on previous studies (Wan et al. 2007; DeRango-Adem et al. 2021). The dose fraction of apigenin converted to A7G (Fm,apigenin→A7G) was estimated to be 0.838 (Eq. 2), indicating that A7G is the major metabolite of apigenin. This result is consistent with those of previous studies reporting that apigenin undergoes glucuronidation to a greater extent than sulfation (Cai et al. 2007). The Vss of A7G was significantly higher than that of apigenin (p = 0.00804). This could be attributed to lower plasma protein binding of A7G compared to that of apigenin (0.010 for apigenin vs. 0.108 for A7G). The dose fraction of A7G converted to apigenin (Fm,A7G→apigenin) was estimated to be 0.00485 (Eq. 2). This suggests that only a minimal fraction of A7G in the systemic circulation can be converted to apigenin in rats.
The present in vitro metabolism studies showed that the hepatic CLint,S9 of apigenin was 18.5-fold higher than its intestinal CLint,S9. After scaling the hepatic and intestinal CLint,S9 in vitro up to the whole organ, the hepatic intrinsic clearance (CLint,H) of apigenin was estimated to be 200 mL/min, and the intestinal intrinsic clearance (CLint,G) was estimated to be 6.42 mL/min (CLint,H/CLint,G = 31.2). A7G is metabolized via hydrolysis mediated by β-glucuronidase that is an acid hydrolytic enzyme expressed in various tissues and body fluids in rats and humans (O’Leary et al. 2001). Approximately one-third of hepatic β-glucuronidase is located in the endoplasmic reticulum (ER) lumen and a little in lysosomes, where it hydrolyzes glucuronide (Swank et al. 1986; Zhu et al. 1996). Considering the stability of A7G in various buffers, it can be suggested that β-glucuronidase-mediated hydrolysis occurred in the liver and intestine. Therefore, when scaling hepatic and intestinal CLint,S9 in vitro up to the whole organ, CLint,H of A7G was estimated to be 12.5 mL/min, and CLint,G was estimated to be 1.23 mL/min (CLint,H/CLint,G = 10.1).
To determine the contributions of the intestine and liver to pre-systemic elimination of apigenin and A7G, EH and EG were estimated from in vitro clearance and/or permeability data using the well-stirred (Eq. 4) and QGut (Eq. 5) models. Based on the hepatic blood flow (QH, 50–80 mL/min/kg) in rats, EH of apigenin and A7G can be estimated to be 0.0246–0.0389 and 0.0167–0.0264, respectively. The effective intestinal permeability (Peff) of apigenin has been reported to be high (0.62 × 10–4 cm/s) in the duodenum and moderate (0.303 × 10–4 cm/s) in the jejunum till colon of rats (Zhang et al. 2012). Therefore, CLperm and EG of apigenin can be estimated to be 0.172–0.620 mL/min and 0.915–0.979, respectively. Assuming that the Peff of A7G is similar to that of apigenin, the EG of A7G can be estimated to be 0.626–0.896. These model-based analyses suggest that apigenin and A7G can be classified as compounds with low EH and moderate-to-high EG in rats. Alternatively, EH and EG of a drug can be calculated using in vivo pharmacokinetic data based on Eqs. 3 and 6 (Yang et al. 2007; Seo et al. 2022a, 2022b). Assuming that apigenin and A7G were stable in the gastrointestinal tract with negligible enterohepatic circulation and reversible metabolic processes, the Fabs of apigenin and A7G were estimated to be 0.658 and 0.900, respectively (Eq. 6). Following intravenous administration of apigenin and A7G, non-renal blood clearance (CLNR) of apigenin and A7G can be calculated to be 12.6 and 9.04 mL/min/kg, respectively, using their Aeu and RB values. Assuming that non-renal systemic elimination of apigenin and A7G occurred only in the liver (CLNR = CLH), EH of apigenin and A7G was estimated to be 0.157–0.251 and 0.113–0.181 (CLH/QH), respectively. Therefore, EG of apigenin and A7G were estimated to be 0.986–0.987 and 0.609–0.954 using Eq. 3. These results indicated low EH and moderate-to-high EG of apigenin and A7G, showing a similar tendency to EH and EG estimated from in vitro data. In light of the discussion above, the fractions of oral dose unabsorbed from the gut and eliminated by the gut and liver before reaching the systemic circulation were estimated to be 40.2–92.9%, 6.51–58.6%, and 0.023–1.28% of the dose, respectively, for apigenin, and 10%, 54.8–85.8%, and 0.472–6.35%, respectively, for A7G. Compared to A7G, intestinal absorption of apigenin seemed to be limited and incomplete, which may be due to its poor solubility and/or luminal stability.
As described earlier, apigenin and A7G were significantly metabolized by both the intestine and liver, and the total metabolic activity was markedly higher (31.2- and 10.1-fold, respectively) in the liver than in the intestine. Thus, it is plausible that the liver had a greater contribution than the intestine to the systemic metabolism of apigenin and A7G. However, in intestinal and hepatic first-pass effects, it is important to note that a compound with a significant CLint,G can be metabolized by the intestine in a route-dependent manner (Han et al. 2022). During the first pass through the intestine, the exposure of a compound to metabolic enzymes can be prolonged and enhanced owing to its relatively slow membrane permeation rate and low luminal protein binding in the intestine (Cho et al. 2014). This could lead to a higher EG when the compound enters the enterocytes from the gut lumen rather than via systemic circulation, which is called route-dependent intestinal elimination (Noh et al. 2019; Seo et al. 2022a, 2022b). For example, many CYP3A substrate drugs undergo considerable intestinal first-pass effects, although the human CYP3A expression in the intestine is only 1% of that in the liver (Yang et al. 2007; Gertz et al. 2010; Seo et al. 2022a, 2022b). This phenomenon agrees well with the present results for apigenin and A7G, which can explain their high EG and low EH, despite considerably higher intrinsic metabolic activity in the liver than in the intestine.
F is one of the most important factors to consider for effective treatment application of phytochemicals. However, despite the various health benefits of phytochemicals, satisfactory efficacy is often not achieved in clinical settings owing to their low F (Selby-Pham et al. 2017; Hu et al. 2023). To enhance the F of phytochemicals, various drug delivery systems such as prodrugs, emulsions, and nanoparticles have been reported (Vinayak et al. 2019; McClements. 2021; Rassu et al. 2023; Zuccari et al. 2023). In the present study, systemic exposure (AUC) of apigenin was markedly higher (8.60–24.7 fold) at the oral dose of A7G than at the oral dose of apigenin. This suggests that A7G, the glucuronide conjugate of apigenin, can act as a natural prodrug to improve the low F of apigenin, which requires further investigation on the clinical relevance of the present rat data. Furthermore, because A7G is a major active metabolite of apigenin, the F and efficacy of A7G also need to be discussed. Based on the IC50 values reported previously, it appears that apigenin has more potent antioxidant and anticancer (via MMP-9) activities than that of A7G (Fathiazad et al. 2011; Crasci et al. 2017; Kashyap et al. 2022) and that A7G has more potent anti-inflammatory, antidiabetic, anti-Alzheimer, and anticancer (via MMP-3, 8, 13) activities than that of apigenin (Cheng et al. 2013; Crasci et al. 2017; Nguyen et al. 2017). Moreover, the systemic exposure of A7G observed after oral administration of apigenin tended to be higher (1.90–12.4 fold) than that after oral administration of A7G. Taken together, these results showed the potential of A7G as more effective oral dosing form that can enhance the F and efficacy of both apigenin and A7G. Our current findings on apigenin can also be applied to other phytochemicals and their phase-II conjugates, including glucuronides and sulfates, which can serve as good alternatives for enhancing the F of poorly absorbed phytochemicals.
Conclusion
We report, for the first time, a simple and sensitive method for simultaneous quantitative analysis of apigenin and its major metabolite A7G in rat plasma using UPLC-MS/MS. Apigenin exhibited pH dependence and limited intestinal luminal stability, whereas A7G remained stable under all tested conditions. The in vivo pharmacokinetic study demonstrated that apigenin had a very low F value and was mainly metabolized to A7G. Notably, systemic exposure of apigenin after oral administration of A7G was markedly higher than that after oral administration of apigenin. Apigenin and A7G were substantially metabolized in both hepatic and intestinal S9 fractions. Based on the well-stirred and QGut model concepts, they could be classified as compounds with low EH and moderate-to-high EG in rats, indicating a greater contribution of the intestine to pre-systemic elimination of both phytochemicals than that of the liver. Taken together, the low F of apigenin could be attributed to its poor stability in the gastrointestinal lumen and extensive intestinal first-pass effect. Moreover, it was improved by oral administration of A7G, demonstrating the potential of A7G as a natural prodrug for enhancing the low F of apigenin.
References
Borges G, Fong RY, Ensunsa JL, Kimball J, Medici V, Ottaviani JI, Crozier A (2022) Absorption, distribution, metabolism and excretion of apigenin and its glycosides in healthy male adults. Free Radic Biol Med 185:90–96
Cai H, Boocock DJ, Steward WP, Gescher AJ (2007) Tissue distribution in mice and metabolism in murine and human liver of apigenin and tricin, flavones with putative cancer chemopreventive properties. Cancer Chemother Pharmacol 60:257–266
Chang SK, Alasalvar C, Shahidi F (2019) Superfruits: phytochemicals, antioxidant efficacies, and health effects - a comprehensive review. Crit Rev Food Sci Nutr 59:1580–1604
Cheng HL, Zhang LJ, Liang YH, Hsu YW, Lee IJ, Liaw CC, Hwang SY, Kuo YH (2013) Antiinflammatory and antioxidant flavonoids and phenols from cardiospermum halicacabum (Dao Di Ling). J Tradit Complement Med 3:33–40
Cho HJ, Kim JE, Kim DD, Yoon IS (2014) In vitro-in vivo extrapolation (IVIVE) for predicting human intestinal absorption and first-pass elimination of drugs: principles and applications. Drug Dev Ind Pharm 40:989–998
Crasci L, Basile L, Panico A, Puglia C, Bonina FP, Basile PM, Rizza L, Guccione S (2017) Correlating in vitro target-oriented screening and docking: inhibition of matrix metalloproteinases activities by flavonoids. Planta Med 83:901–911
Cui Z, Zhao X, Amevor FK, Du X, Wang Y, Li D, Shu G, Tian Y, Zhao X (2022) Therapeutic application of quercetin in aging-related diseases: SIRT1 as a potential mechanism. Front Immunol 13:943321
Derango-Adem EF, Blay J (2021) Does oral apigenin have real potential for a therapeutic effect in the context of human gastrointestinal and other cancers? Front Pharmacol 12:681477
Ding Y, Peng M, Zhang T, Tao JS, Cai ZZ, Zhang Y (2013) Quantification of conjugated metabolites of drugs in biological matrices after the hydrolysis with beta-glucuronidase and sufatase: a review of bio-analytical methods. Biomed Chromatogr 27:1280–1295
Elzayat EM, Shakeel F, Alshehri S, Ibrahim MA, Altamimi MA, Kazi M, Alanazi FK, Haq N (2019) UHPLC assisted simultaneous separation of apigenin and prednisolone and its application in the pharmacokinetics of apigenin. J Chromatogr B Analyt Technol Biomed Life Sci 1117:58–65
Fathiazad F, Mazandarani M, Hamedeyazdan S (2011) Phytochemical analysis and antioxidant activity of Hyssopus officinalis L. from Iran. Adv Pharm Bull 1:63–67
Fraga CG, Croft KD, Kennedy DO, Tomas-Barberan FA (2019) The effects of polyphenols and other bioactives on human health. Food Funct 10:514–528
Gao S, Sun R, Singh R, Yu So S, Chan CTY, Savidge T, Hu M (2022) The role of gut microbial beta-glucuronidase in drug disposition and development. Drug Discov Today 27:103316
Gertz M, Harrison A, Houston JB, Galetin A (2010) Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab Dispos 38:1147–1158
Gontijo VS, Dos Santos MH, Viegas C Jr (2017) Biological and chemical aspects of natural biflavonoids from plants: a brief review. Mini Rev Med Chem 17:834–862
Gorzynik-Debicka M, Przychodzen P, Cappello F, Kuban-Jankowska A, Marino Gammazza A, Knap N, Wozniak M, Gorska-Ponikowska M (2018) Potential health benefits of olive oil and plant polyphenols. Int J Mol Sci 19:686
Gradolatto A, Canivenc-Lavier MC, Basly JP, Siess MH, Teyssier C (2004) Metabolism of apigenin by rat liver phase I and phase II enzymes and by isolated perfused rat liver. Drug Metab Dispos 32:58–65
Gradolatto A, Basly JP, Berges R, Teyssier C, Chagnon MC, Siess MH, Canivenc-Lavier MC (2005) Pharmacokinetics and metabolism of apigenin in female and male rats after a single oral administration. Drug Metab Dispos 33:49–54
Han DG, Seo SW, Choi E, Kim MS, Yoo JW, Jung Y, Yoon IS (2022) Impact of route-dependent phase-II gut metabolism and enterohepatic circulation on the bioavailability and systemic disposition of resveratrol in rats and humans: a comprehensive whole body physiologically-based pharmacokinetic modeling. Biomed Pharmacother 151:113141
Hu Y, Lin QZ, Zhao H, Li XJ, Sang SY, Mcclements DJ, Long J, Jin ZY, Wang JP, Qiu C (2023) Bioaccessibility and bioavailability of phytochemicals: influencing factors, improvements, and evaluations. Food Hydrocoll 135:108165
Imran M, Aslam Gondal T, Atif M, Shahbaz M, Batool Qaisarani T, Hanif Mughal M, Salehi B, Martorell M, Sharifi-Rad J (2020) Apigenin as an anticancer agent. Phytother Res 34:1812–1828
Iwaki M, Ogiso T, Ito Y (1989) Pharmacokinetics and biotransformation of hydralazine acetone hydrazone, a metabolite of hydralazine, in the rat. J Pharm Sci 78:867–873
Jin T, Chi L, Ma C (2022) Agrimonia pilosa: a phytochemical and pharmacological review. Evid Based Complement Alternat Med 2022:3742208
Kashyap P, Shikha D, Thakur M, Aneja A (2022) Functionality of apigenin as a potent antioxidant with emphasis on bioavailability, metabolism, action mechanism and in vitro and in vivo studies: a review. J Food Biochem 46:e13950
Keem M-J, Seo S-W, Kim T, Jo B-G, Kim S-N, Yoon I-S, Yang MH (2023) A high-performance liquid chromatography with photodiode array detection method for simultaneous determination of three compounds isolated from Wikstroemia ganpi: assessment of the effects on cytochrome P450-mediated metabolism in vitro and in vivo. Nutrients 15:4061
Kumar S, Pandey AK (2013) Chemistry and biological activities of flavonoids: an overview. ScientificWorldJournal 2013:162750
Kurnia D, Putri SA, Tumilaar SG, Zainuddin A, Dharsono HDA, Amin MF (2023) In silico study of antiviral activity of polyphenol compounds from ocimum basilicum by molecular docking, ADMET, and drug-likeness analysis. Adv Appl Bioinform Chem 16:37–47
Li L, Jiang H, Wu H, Zeng S (2005) Simultaneous determination of luteolin and apigenin in dog plasma by RP-HPLC. J Pharm Biomed Anal 37:615–620
Liu Y, Hu M (2002) Absorption and metabolism of flavonoids in the caco-2 cell culture model and a perused rat intestinal model. Drug Metab Dispos 30:370–377
Marrassini C, Cogoi L, Sulsen V, Anesini C (2020) Apigenin-7-Glucuronide from urera aurantiaca inhibits tumor necrosis factor alpha and total nitrite release in lipopolysaccharide-activated macrophages. Evid Based Complement Alternat Med 2020:6638764
Mcclements DJ (2021) Advances in edible nanoemulsions: digestion, bioavailability, and potential toxicity. Prog Lipid Res 81:101081
Nguyen DH, Seo UM, Zhao BT, Le DD, Seong SH, Choi JS, Min BS, Woo MH (2017) Ellagitannin and flavonoid constituents from Agrimonia pilosa Ledeb. With their protein tyrosine phosphatase and acetylcholinesterase inhibitory activities. Bioorg Chem 72:293–300
Noh K, Pang KS (2019) Theoretical consideration of the properties of intestinal flow models on route-dependent drug removal: segregated Flow (SFM) vs. traditional (TM). Biopharm Drug Dispos 40:195–213
O’leary KA, Day AJ, Needs PW, Sly WS, O’brien NM, Williamson G (2001) Flavonoid glucuronides are substrates for human liver β-glucuronidase. FEBS Lett 503:103–106
Prochazkova D, Bousova I, Wilhelmova N (2011) Antioxidant and prooxidant properties of flavonoids. Fitoterapia 82:513–523
Rana A, Samtiya M, Dhewa T, Mishra V, Aluko RE (2022) Health benefits of polyphenols: a concise review. J Food Biochem 46:e14264
Rassu G, Sorrenti M, Catenacci L, Pavan B, Ferraro L, Gavini E, Bonferoni MC, Giunchedi P, Dalpiaz A (2023) Conjugation, prodrug, and co-administration strategies in support of nanotechnologies to improve the therapeutic efficacy of phytochemicals in the central nervous system. Pharmaceutics 15:1578
Salehi B, Venditti A, Sharifi-Rad M, Kregiel D, Sharifi-Rad J, Durazzo A, Lucarini M, Santini A, Souto EB, Novellino E, Antolak H, Azzini E, Setzer WN, Martins N (2019) The therapeutic potential of apigenin. Int J Mol Sci 20:1305
Selby-Pham SNB, Miller RB, Howell K, Dunshea F, Bennett LE (2017) Physicochemical properties of dietary phytochemicals can predict their passive absorption in the human small intestine. Sci Rep 7:1931
Seo SW, Han DG, Choi E, Seo MJ, Song IS, Yoon IS (2022a) Factors determining the oral absorption and systemic disposition of zeaxanthin in rats: in vitro, in situ, and in vivo evaluations. Pharm Biol 60:2266–2275
Seo SW, Han DG, Choi E, Park T, Byun JH, Cho HJ, Jung IH, Yoon IS (2022b) Development and application of a physiologically based pharmacokinetic model for entrectinib in rats and scale-up to humans: route-dependent gut wall metabolism. Biomed Pharmacother 146:112520
Seo SW, Han DG, Baek YM, Park MC, Yoo JW, Jung Y, Maeng HJ, Myung H, Yoon IS (2023) Investigation of the factors responsible for the low oral bioavailability of alizarin using a sensitive LC-MS/MS method: in vitro, in situ, and in vivo evaluations. Drug Dev Res 84:579–591
Seong GS, Seo SW, Cho JY, Lee KW, Lee BJ, Yoon IS, Jin HE (2022) Determination of leuprolide-fatty acid conjugate in rat plasma using LC-MS/MS and its pharmacokinetics after subcutaneous administration in rats. Molecules 27:8716
Serafini M, Peluso I, Raguzzini A (2010) Flavonoids as anti-inflammatory agents. Proc Nutr Soc 69:273–278
Shia CS, Hou YC, Tsai SY, Huieh PH, Leu YL, Chao PD (2010) Differences in pharmacokinetics and ex vivo antioxidant activity following intravenous and oral administrations of emodin to rats. J Pharm Sci 99:2185–2195
Swank RT, Pfister K, Miller D, Chapman V (1986) The egasyn gene affects the processing of oligosaccharides of lysosomal beta-glucuronidase in liver. Biochem J 240:445–454
Tang L, Zhou J, Yang CH, Xia BJ, Hu M, Liu ZQ (2012) Systematic studies of sulfation and glucuronidation of 12 flavonoids in the mouse liver S9 fraction reveal both unique and shared positional preferences. J Agric Food Chem 60:3223–3233
Trontelj J (2012) Quantification of glucuronide metabolites in biological matrices by LC-MS/MS. Tandem Mass Spectrom Appl Princ 531–558
Tu Y, Zhou L, Li L, Wang L, Gao S, Hu M (2020) Development and validation of an LC-MS/MS method for the quantification of flavonoid glucuronides (wogonoside, baicalin, and apigenin-glucuronide) in the bile and blood samples: application to a portal vein infusion study. Anal Biochem 601:113723
US Food and Drug Administration (2018) Bioanalytical Method Validation Guidance for Industry. https://www.fda.gov/media/70858/download. Accessed 16 May 2023
Vinayak M, Maurya AK (2019) Quercetin loaded nanoparticles in targeting cancer: recent development. Anticancer Agents Med Chem 19:1560–1576
Vo D-K, Nguyen T-T-L, Maeng H-J (2022) Effects of 1α, 25-dihydroxyvitamin D3 on the pharmacokinetics and biodistribution of ergothioneine, an endogenous organic cation/carnitine transporter 1 substrate, in rats. J Pharm Investig 52:341–351
Vucic V, Grabez M, Trchounian A, Arsic A (2019) Composition and potential health benefits of pomegranate: a review. Curr Pharm Des 25:1817–1827
Wan L, Guo C, Yu Q, Li Y, Wang X, Wang X, Chen C (2007) Quantitative determination of apigenin and its metabolism in rat plasma after intravenous bolus administration by HPLC coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 855:286–289
Wang MQ, Firrman J, Liu LS, Yam K (2019) A review on flavonoid apigenin: dietary intake, ADME, antimicrobial effects, and interactions with human gut microbiota. Biomed Res Int 2019:7010467
Wang Y, Jia M, Gao Y, Zhao B (2023) Multiplex quantitative analysis of 9 compounds of Scutellaria baicalensis georgi in the plasma of respiratory syncytial virus-infected mice based on HPLC-MS/MS and pharmacodynamic effect correlation analysis. Molecules 28:6001
Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A (2007) Prediction of intestinal first-pass drug metabolism. Curr Drug Metab 8:676–684
Yoon IS, Choi MK, Kim JS, Shim CK, Chung SJ, Kim DD (2011) Pharmacokinetics and first-pass elimination of metoprolol in rats: contribution of intestinal first-pass extraction to low bioavailability of metoprolol. Xenobiotica 41:243–251
Yun T-S, Jung M, Bang K-H, Lee H-K, Jin M, Yoo H, Won J-H, Song B, Hwang Y-R, Baek J-S (2023) An economically advantageous amorphous solid dispersion of the fixed combination of lopinavir and ritonavir. J Pharm Investig 53:549–561
Zhang J, Liu D, Huang Y, Gao Y, Qian S (2012) Biopharmaceutics classification and intestinal absorption study of apigenin. Int J Pharm 436:311–317
Zhu BT, Evaristus EN, Antoniak SK, Sarabia SF, Ricci MJ, Liehr JG (1996) Metabolic deglucuronidation and demethylation of estrogen conjugates as a source of parent estrogens and catecholestrogen metabolites in Syrian hamster kidney, a target organ of estrogen-induced tumorigenesis. Toxicol Appl Pharmacol 136:186–193
Zhu S, Zhang J, Lv Z, Yu M (2021) LC-MS/MS determination of apigenin in rat plasma and application to pharmacokinetic study. Curr Pharm Biotechnol 22:274–280
Zuccari G, Alfei S (2023) Development of phytochemical delivery systems by nano-suspension and nano-emulsion techniques. Int J Mol Sci 24:9824
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea Government (MSIT) (NRF-2023R1A2C1006010) and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI23C0141).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors (S.W. Seo, S.H. Choi, J.K. Hong, K.M. Kim, S.C. Kang and I.S. Yoon) declare that they have no conflict of interest.
Research involving human and animal rights
Animal studies were performed according to the protocol approved by Institutional Animal Care and Use Committee of Pusan National University (approval number: PNU-2023-3245; Busan, Republic of Korea).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Seo, SW., Choi, S.H., Hong, JK. et al. Pharmacokinetics and extensive intestinal first-pass effects of apigenin and its active metabolite, apigenin-7-O-glucuronide, in rats. J. Pharm. Investig. 54, 467–481 (2024). https://doi.org/10.1007/s40005-024-00662-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40005-024-00662-1