
Abstract
Liposomes have been long used in cancer therapy with great expectations that they can improve efficacy mainly through enhanced permeation and retention (EPR) effect and reduce toxicity related to un-encapsulated anticancer agents. However, the advantage of liposomal formulations has not been prominent as anticipated in clinical cancer treatment in contrast to preclinical results and this has led nanomedicine field to review the experiences of liposomal anticancer products so far to pursue better strategies. The aim of this review is to look for answers to the questions whether liposomes really increase efficacy and reduce toxicity of encapsulated drugs in humans by consulting recent several meta-analyses. Based on the meta-analyses, liposomal formulations have shown comparable or modestly superior clinical efficacy compared to non-liposomal conventional formulations. Besides, drug-related toxicity, for example, cardiotoxicity of doxorubicin, has been clearly reduced by encapsulation of cytotoxic agents into liposomes despite carrier-related adverse events are newly occurred. In conclusion, liposomes are clinically useful in cancer therapy and their value can be raised by applying more advanced and elaborate strategies through understanding tumor microenvironment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since Bangham et al. reported the discovery of liposome in Bangham et al. (1965), liposome has drawn great attentions as drug carrier for conventional drugs due to its versatile nature and it took almost three decades to be approved for clinical use. In 1995, a sterically stabilized liposomal formulation for doxorubicin, Doxil®, was approved by FDA for the treatment of AIDS-related Kaposi’s sarcoma and its indications has been extended to breast cancer, ovarian cancer and multiple myeloma (Gabizon et al. 2016). Five years later, a non-PEGylated liposomal formulation for doxorubicin, Myocet®, was approved in the EU and Canada. These successes have thrown optimism to many researchers and companies and led extensive tries to develop liposomal formulations for various conventional drugs. In many cases, liposome has been tried as a delivery platform of choice to modify pharmacokinetics and solve formulation difficulties of free drugs in life-threatening diseases such as cancer due to its biocompatibility and versatile physico-chemical properties.
One of the reasons behind these successes and optimistic prospects has been suggested to be the Enhanced Permeation and Retention (EPR) effect by which drugs could be passively directed to the tumor sites with leaky vasculature. EPR was first reported in the treatment of hepatocarcinomas with SMANCS system by Maeda group and expanded upon other macromolecular and nano-sized drug delivery systems (Maeda et al. 2016). The high permeable vasculature in tumor allows enhanced permeation for relatively large particles such as proteins, macromolecules, liposomes, micelles and other particles into the interstitial space of the tumor consisted of impaired lymphatic drainage limiting the clearance of particles from tumor and consequently causing enhanced retention of the particles (Maeda et al. 2016).
However, there have been reports that nanomedicine including liposomes did not show EPR effect in many clinical cancers contrary to the preclinical results (Nichols and Bae 2014). In addition, some pharmaceutical investigators might hesitate to try liposome as an alternative formulation for their anticancer drugs because liposomes usually require more efforts and costs in the way from the laboratory bench to clinical trials or to the market compared to other conventional formulation techniques (Satalkar et al. 2016; Hare et al. 2017). Despite of these doubts, several other liposomal products have been approved (Table 1) and under clinical trials at different stages (Table 2), which indicates that liposomes still have great potential as drug delivery system. In this regard, there has been demands to prove the usefulness or advantage of liposomal formulations over conventional ones especially in clinical applications. Fortunately, thanks to several meta-analysis studies, it is possible to some extent to answer to questions asking that liposomes can really improve efficacy and reduce toxicity.
The aim of this review is to provide comprehensive overview regarding clinical usefulness of liposomal formulations by focusing on the several meta-analyses for liposomal doxorubicin and deliver hopeful message that liposome is worth a try for sake of patients.
Liposomal formulations for doxorubicin
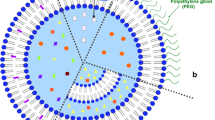
Doxorubicin-containing stealth liposomal product was commercialized in the US as Doxil® and in the EU as Caelyx®. As shown in Fig. 1, it consists PEGylated liposomal bilayer with a size of 80–90 nm and the composition of bilayer is hydrogenated soy phosphatidylcholine (HSPC), cholesterol (CHOL) and methyl-distearoyl phosphor-ethanolamine PEG2000 (DSPE-PEG2000) sodium salt in a molar ratio of 56:39:5 (Barenholz 2012). Originally, the researchers designed non-PEGylated oligo-lamellar liposomal formulation (OLV-DOX) to fail in a clinical trial because rapid release of drug from liposomes in plasma resulting in cardiotoxicity of doxorubicin, fast elimination from the circulation by reticuloendothelial system (RES) and inhibited extravasation by the large size (200–500 nm). To overcome the limitations of OLV-DOX, PEGylated phospholipid was introduced and remote loading technique applied. The addition of PEG polymers enabled liposomes to stay for extended period in the circulation by avoiding RES recognition, which in turn has been believed to result in sufficient accumulation in tumor site through EPR effect (Yingchoncharoen et al. 2016). In addition, remote-loading technology allowed encapsulation efficiency of doxorubicin higher than 90% and most doxorubicin to be trapped inside liposomes during circulation.

Schematic illustration of liposomal doxorubicin
After the success of Doxil®, a non-PEGylated liposomal doxorubicin product, named Myocet®, was approved by the EMA and Health Canada. Myocet® liposome consists of egg PC and cholesterol in a molar ratio of 55:45 and the encapsulation of doxorubicin is carried out by remote loading similar to Doxil®. However, Myocet® used a pH gradient rather than ammonium sulfate gradient as a driving force for the remote loading of doxorubicin into liposomes formed beforehand (Yingchoncharoen et al. 2016). Myocet® shows encapsulation efficiency higher than 95% and liposomal size of approximately 150 nm (Fig. 1).
There were no direct comparative studies between Doxil® and Myocet® in humans to date which may be because two products exhibit remarkably different pharmacokinetics of doxorubicin. Even though it is difficult to determine a comparable effective dose for each formulation, they have been used with different dosing regimens and compared indirectly through a meta-analysis (Petersen et al. 2016). As expected, patients have been usually treated with lower dose of Doxil® (40–50 mg/m2 in every 21 or 28 days) than Myocet® (60–75 mg/m2 in every 21 days) (Batist et al. 2001; Dimopoulos et al. 2003; Harris et al. 2002; Hunault-Berger et al. 2011; Judson et al. 2001; O’Brien et al. 2004; Rifkin et al. 2006). A meta-analysis selected clinical trials comparing liposomal and non-liposomal anthracyclines and showed no significant advantage in clinical efficacy in liposomal formulations over non-liposomal conventional ones regardless of PEGylated or non-PEGylated (Petersen et al. 2016). However, Doxil® and Myocet® present clearly different toxicity profile each other (Yingchoncharoen et al. 2016). The two products are also provided as different dosage forms as shown in Table 3; liposomal dispersion for Doxil® which can be used directly only after dilution with 5% dextrose solution, while separate three vials for Myocet® containing lyophilized free drug powder, pre-formed empty liposomal dispersion and buffer solution which should be mixed with heating under aseptic condition for free drug to be encapsulated into liposomes remotely and then diluted with 0.9% NaCl or 5% dextrose solution before use. As we have seen in Myocet®, it appears to be still quite a challenge to prepare each-to-use nano-sized liposomal dosage form for intravenous application because of some limitations such as release of encapsulated drugs and particle size change during storage.
Comparable clinical efficacy of liposomal formulations
Nanoparticles including liposomes have been regarded as promising drug delivery systems for cytotoxic drugs because they can increase drug delivery to tumor sites while limiting drug distribution to normal tissues especially from the EPR effect (von Roemeling et al. 2017). However, drug carriers generally failed to provide superior efficacy to free drug when tested in clinical trials (Nichols and Bae 2014). In spite of the downfall of the EPR effect in clinical settings, liposomes have been long used in cancer treatment and still tried as delivery system for several anticancer agents as shown in Table 2. In this regard, researchers saw the necessity to address the fundamental question whether liposomal formulations significantly increase the efficacy of drugs compared to conventional non-liposomal formulations (Petersen et al. 2016; Yamaguchi et al. 2015; Xing et al. 2015).
Petersen et al. performed a meta-analysis using clinical trials published from 1990 to 2015 comparing liposomal and non-liposomal formulations for anticancer agents (Petersen et al. 2016). Based on their inclusion criteria, they selected 14 randomized clinical trials directly comparing efficacy of liposomal drug and their equivalent non-liposomal drug (8 anthracyclines, 4 cisplatin, 1 paclitaxel, 1 irinotecan) as shown in Table 4. The differences of efficacy between formulations were analyzed by comparing objective response rate (ORR), progression-free survival (PFS) and overall survival (OS). According to their analysis, there were no significant differences of ORR, PFS and OS between liposomes and non-liposomal formulations. They also performed subgroup analysis using eight anthracycline trials. In contrast to our common expectation, liposomal anthracyclines showed no significant difference in ORR, PFS and OS from the equivalent non-liposomal ones. Meanwhile, platinum-liposomes showed significant increase in ORR compared to free cisplatin even though OS and PFS were not analyzed due to the lack of the number of studies reporting these endpoints. Petersen et al. noticed that all of four cisplatin trials were for non-small cell lung carcinoma and thus performed subgroup analysis for metastatic breast cancer to see tumor-type dependency. However, the subgroup analysis for three metastatic breast cancer trials did not show any improvement of efficacy by liposomal formulations of anthracyclines (1 PEGylated liposome, 2 conventional liposomes). They also failed to show advantage of liposomes in the anticancer efficacy of anthracyclines regardless of PEGylated or non-PEGylated in the comparison of PEGylated liposomal versus free anthracyclines (5 trials) and non-PEGylated liposomal versus free anthracyclines (3 trials). They suggested that the moderate improvement of efficacy in cisplatin liposomes may be related to the drug encapsulated (i.e., cisplatin) or cancer type (i.e., lung cancer) because tumor-related parameters such as vascularity can have significant impact on the pharmacokinetics of liposomal drugs.
Another meta-analysis study selected 10 clinical trials for liposomal doxorubicin reported until April 2015 as shown in Table 5 (Xing et al. 2015). The meta-analysis showed liposomal doxorubicin improved ORR significantly, though no significant difference in PFS and OS compared to non-liposomal doxorubicin. In contrast to the analysis by Petersen et al., Xing et al. included the studies that free and encapsulated drugs were not the same one.
Based on the meta-analyses, liposomal formulations for anticancer agents have shown comparable clinical efficacy to the un-encapsulated conventional formulations contrary to our expectations that liposomes would show superior anticancer efficacy mainly due to the EPR effect. EPR effect of nanoparticles has been demonstrated in a lot of animal experiments and led superior efficacy of nanoparticles (Fang et al. 2011). Thus, Petersen et al. also performed a meta-analysis of pre-clinical studies comparing efficacy of PEGylated liposomal formulations and conventional ones for doxorubicin (Petersen et al. 2016). They selected 11 studies (Table 6) in which both PEGylated liposomal and conventional doxorubicin were contained, anticancer activity evaluated in at least one tumor model and the survival curves reported. On the contrary to the meta-analysis results for clinical trials, PEGylated liposomal doxorubicin showed significantly improved survival rate in mice than conventional doxorubicin (Petersen et al. 2016). Recent studies have found that empty nanoparticles including liposomes enhanced tumor growth in tumor-bearing immuno-competent mice (Moghimi 2014; Sabnani et al. 2015). This was associated with increased tumor angiogenesis and suppression of antitumor immune response in immuno-competent mice (Sabnani et al. 2015). Based on these findings, Petersen et al. noticed immuno-microenvironment could be one of explanations for the lack of clinical advantage of liposomes. They performed sub-group analysis of immuno-competent (n = 5) and immuno-deficient (n = 4) mice models and showed the superior efficacy of liposomes was also demonstrated in both sub-groups. They seemed to predict that immuno-competent mice sub-group would show no advantage of liposomes. However, immune-competent subgroups also exhibited superiority of liposomes and they thought the tumor-promoting effect of liposomes would not likely to be detected in their meta-analysis because 5 of the 11 preclinical studies used immune-deficient athymic nude mice and even wild-type mouse strain such as DBA/2 can have observable immuno-defects due to inbreeding. They pointed out that most preclinical studies have not reported immune assessment for liposomes and other nanoparticles although nanoparticles have been known to have various immunological effects (Ilinskaya and Dobrovolskaia 2014; Dobrovolskaia et al. 2009).
In addition to the immuno-environment, several plausible explanations have been suggested for the failure of translation of the superior efficacy of liposomes obtained from EPR effect in pre-clinical studies into enhanced clinical results. First, we can consider limitations of animal models. Liposomal anticancer agents are usually used for patients with advanced metastatic cancers which have been developed over a very long period. However, most murine tumor models are xenograft which grows fast and the experiments are finished within several weeks. Due to the rapid growth, angiogenesis rate can be higher in murine tumor model than human cancer which in turn can show exaggerated EPR effect (Danhier 2016; Petersen et al. 2016; Fang et al. 2011). Besides, human cancers show genetic diversity because they should survive against immune system for a long time and thus are difficult to treat (Choi et al. 2011; Nichols and Bae 2014). On the other hand, murine tumor cells grow relatively free from immune pressure for short period and thus result in genetically less diverse cells compared to clinical human tumors and tumors with under-developed secondary structures such as pericytes, basement membrane, and extracellular matrix (Danhier 2016; Nichols and Bae 2014). Another possible explanation is that most pre-clinical studies have reported tumor size as efficacy endpoint instead of survival curve which is regarded as a gold-standard measurement of efficacy in clinical trials (Petersen et al. 2016). Tumor response rates such as size shrinkage are not considered as accurate predictors for survival benefit in clinical trials (Johnson et al. 2006). Pre-clinical efficacy would be more accurately translated into clinical effectiveness when survival is used as an efficacy endpoint in animal studies. Dosing regimen can be one of the reasons causing inconsistency between pre-clinical and clinical results. Most clinical dosing schedules for anticancer agents are designed for conventional formulations rather than for liposomal ones even though it has been well-known that encapsulation of drugs into carriers can cause marked changes in pharmacokinetics and distribution of drugs (Petschauer et al. 2015). Moreover, tumor-to-body weight ratio is much higher in murine tumor model compared to human cancer and this can cause significant difference of pharmacokinetics between animal models and human patients. In murine model, tumor size can reach as much as 10% of the body weight and thus significant portion of administered dose can be deposited in tumor to show improved efficacy (Nichols and Bae 2014). Besides, more aggressive dosing schedule has been applied in animal models compared to human. In the treatment schedule for human patients, 2–4 weeks dosing intervals are used to allow for the patients to recover from toxic effect of anticancer drugs and this might be able to give a chance to cancer cells to recover or turn into resistant cells (Nichols and Bae 2014).
Finally, we need to consider variation of pharmacodynamics resulted from high inter-subject pharmacokinetic variability of liposomal anticancer agents. Shell et al. showed liposomal formulations demonstrated significantly higher inter-subject variation in pharmacokinetic parameters compared to non-liposomal formulations (Schell et al. 2014). They analyzed commercially available or under-developing nine liposomal formulations as shown in Table 7. Liposomal formulations showed threefold higher inter-subject variation in AUC compared to non-liposomal formulations, and there was a case that showed even 10-fold difference in AUC (Schell et al. 2014). High pharmacokinetic variability will result in large deviation in drug exposure and finally cause inter-subject variation in efficacy. The effect can be prominent in the case of anticancer agents with narrow therapeutic window. Schell et al. suggested mononuclear phagocytic system (MPS) would be the most dominant factor to affect pharmacokinetic variability of liposomes. According to the meta-analysis, liposomal formulations with lower systemic clearance showed a tendency to lead to higher inter-subject variation of AUC, while no such tendency in free anticancer drugs (Schell et al. 2014). In case of CKD602, increasing dose of liposomes reduced pharmacokinetic variations and they suggested this could be due to the saturation of liposome clearance by MPS. In contrast to liposomes, free CKD602 showed no correlation between dose and pharmacokinetic variability. From this results, Schell et al. suggested that extended circulation time of PEGylated liposomes would increase variability of pharmacokinetics (PK) and pharmacodynamics (PD). However, two other PEGylated liposomes such as Doxil® and IHL-305 exhibited lower variability compared to PEGylated liposomal formulation of CKD602 (S-CKD602). In these aspects, we need to pay attention to the non-bioequivalence of Lipodox® (Sun Pharmaceutical Inc., Mumbai, India) approved as a generic for Doxil® although there was no clear study on the reason for the non-bioequivalence (Yingchoncharoen et al. 2016). According to the clinical trial for ovarian cancer, Doxil® showed 18% of ORR and 7.2 months of PFS while Lipodox only 4.3% of ORR and 5.4 months of PFS. On the whole, Lipodox® showed approximately 15.7–21.3% lower activity compared to Doxil® and this was likely due to the lower accumulation of Lipodox® into tumor (Yingchoncharoen et al. 2016). Considering high variability in PK/PD of liposomal formulations and the difference between the generic and original products, behavior of liposomes would be altered responding very sensitively to the slight changes such as particle size and drug release rate. Consequently, the high sensitivity results in large variations in clinical efficacy which in turn make it difficult to detect advantage of liposomes over conventional formulations in clinical situations. In this aspect, it would be also important to prepare homogeneous with narrow particle size distribution and reproducible liposomes to achieve improved clinical effectiveness compared to conventional formulations.
Reduced toxicity by liposomal formulations
Nano-carriers including liposomes have been regarded as delivery systems to reduce adverse effects of free cytotoxic drugs preserving anticancer activities (Brand et al. 2017). Especially, anthracycline-containing liposomes have been acknowledged for reduced occurrence of cardiotoxicity compared to conventional formulations (Cagel et al. 2017). Anthracyclines are one of the most effective and first-line treatments in many forms of cancers including breast cancer and lymphoma. However, the benefits of anthracyclines are restricted by the risk of cardiotoxicity which is known to be related to cumulative dose (Dong and Chen 2018). Several studies have shown that liposomal doxorubicin reduced or did not increase cardiotoxicity compared to conventional doxorubicin or anthracycline-free chemotherapy (Olivieri et al. 2017; Batist et al. 2001; Harris et al. 2002; Sparano et al. 2009). On the other hand, some trials reported that liposomal doxorubicin presented similar cardiotoxicity with higher effectiveness than conventional formulation (Chan et al. 2004). The inconsistency have led Xing et al. to conduct a meta-analysis by combining results from all eligible randomized controlled trials listed in Table 5 (Xing et al. 2015). They pooled data from 2889 advanced breast cancer patients in ten trials in Table 4 and concluded that cardiotoxicity appeared to occur less frequently in patients treated with liposomal doxorubicin-based chemotherapy compared with conventional doxorubicin. They commented that their study was the first meta-analysis to their knowledge which combined the data of existing published studies and thus has reduced the effect of published bias. Xing et al. also acknowledged a few limitations of their meta-analysis; only cardiotoxicity was considered and the definition of cardiotoxicity they used was based on significant left ventricular ejection fraction (LVEF) changes which was not uniform across all trials (Xing et al. 2015). In spite of the limitations, the meta-analysis result is encouraging in terms of reducing toxicity and thus liposomal doxorubicin can be a clinically advantageous alternative in chemotherapy.
Another meta-analysis was performed to compare cardiac events associated liposomal doxorubicin and free anthracyclines such as epirubicin and doxorubicin in breast cancer (Yamaguchi et al. 2015). Yamaguchi et al. reviewed randomized controlled trials evaluating either regimens in the metastatic or adjuvant settings published up to January of 2014 and selected 19 trials which allowed direct and indirect comparison of cardiotoxicity of different anthracyclines and non-anthracyclines. Six studies out of the 19 trials included liposomal formulations as shown in Table 8 and all of them were also contained in the meta-analysis by Xing et al. (Xing et al. 2015). According to the results by the meta-analysis, conventional doxorubicin showed higher cardiac events grade 3 or greater (CE3) than non-anthracycline-based regimen and other anthracycline, epirubicin (Yamaguchi et al. 2015). Liposomal doxorubicin tended to lower cardiotoxicity than free doxorubicin with statistical significance. However, liposomal doxorubicin did not present advantage over epirubicin and non-anthracycline by showing no statistically significant differences in respect of cardiotoxicity. Yamaguchi et al. suggested that anthracyclines can be safely used as far as they do not exceed the cumulative dose limit because the incidence of CE3 was generally low in any type of anthracyclines. Even though CE3 does not limit the choice of anthracycline, we need to pay attention to the result that liposomal doxorubicin appeared to be the least cardiotoxic formulation among the compared formulations such as non-anthracyclines, free doxorubicin, epirubicin and liposomal doxorubicin.
The two meta-analyses taken together, we can conclude that liposomal formulations significantly reduce the toxicity of un-encapsulated conventional cytotoxic drugs. Even though liposomes can provide advantage in reducing toxicity of free drug, encapsulation of cytotoxic drugs sometimes resulted in a change of toxicological profile and thus produce other adverse reactions which has not been observed in conventional formulations. For example, PEGylated liposomes containing doxorubicin (Doxil®) produce skin reactions known as hand and foot syndrome or palmar plantar erythro-dysthesia (PPE) which is related to the extended circulation time (Yingchoncharoen et al. 2016). Due to the long circulation time and low clearance rate, Doxil® can penetrate into skin tissue readily, making it more effective for the treatment of AIDS-related Kaposi sarcoma and also resulting in PPE as a side effect. On the other hand, PPE rarely occurs in non-PEGylated liposomes of doxorubicin (Myocet®) (Yingchoncharoen et al. 2016). However, Myocet® induces an increased bone marrow suppression than free doxorubicin formulation (Brand et al. 2017).
Lower incidence of API-related toxicity by liposomal formulations has been reported also for drugs other than cytotoxic agents (Brand et al. 2017). Encapsulation of amphotericin B into liposomes reduced adverse events of nephrotoxicity associated with amphotericin B (Steimbach et al. 2017; Brand et al. 2017). According to a meta-analysis for amphotericin B formulations, all lipid formulations (liposomes, lipid complex, colloidal dispersion and Intralipid infusion) presented better profiles than the conventional formulation with respect to the adverse events of nephrotoxicity, fever, chills and vomiting, while exhibited the same efficacy profile with conventional formulation (Steimbach et al. 2017).
Conclusion
It appeared to be encouraging that meta-analyses showed liposomal formulations are comparable or modestly superior in clinical efficacy compared to non-liposomal conventional formulations despite of some doubts related to the ambiguous EPR effect in humans. Besides, drug-related toxicity, for example, cardiotoxicity of doxorubicin, has been clearly reduced by encapsulation of cytotoxic agents into liposomes despite carrier-related adverse events are newly occurred. As many researchers indicated, EPR effect extremely heterogeneous in humans and thus it would be necessary to stop claiming enhanced efficacy via EPR effect. It requires to develop alternative strategies which do not depend on EPR effect to raise clinical value of liposomes in chemotherapy. Otherwise, more elaborate pre-clinical study should be considered to provide meaningful prediction of efficacy in humans.
References
Anders CK, Adamo B, Karginova O, Deal AM, Rawal S, Darr D, Schorzman A, Santos C, Bash R, Kafri T, Carey L, Miller CR, Perou CM, Sharpless N, Zamboni WC (2013) Pharmacokinetics and efficacy of PEGylated liposomal doxorubicin in an intracranial model of breast cancer. PLoS One 8(5):e61359
Bangham AD, Standish MM, Watkins JC (1965) Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol 13(1):238–252
Barenholz Y (2012) Doxil®(R)--the first FDA-approved nano-drug: lessons learned. J Control Release 160(2):117–134
Baselga J, Manikhas A, Cortes J, Llombart A, Roman L, Semiglazov VF, Byakhov M, Lokanatha D, Forenza S, Goldfarb RH, Matera J, Azarnia N, Hudis CA, Rozencweig M (2014) Phase III trial of nonPEGylated liposomal doxorubicin in combination with trastuzumab and paclitaxel in HER2-positive metastatic breast cancer. Ann Oncol 25(3):592–598
Batist G, Ramakrishnan G, Rao CS, Chandrasekharan A, Gutheil J, Guthrie T, Shah P, Khojasteh A, Nair MK, Hoelzer K, Tkaczuk K, Park YC, Lee LW (2001) Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J Clin Oncol 19(5):1444–1454
Brand W, Noorlander CW, Giannakou C, De Jong WH, Kooi MW, Park MV, Vandebriel RJ, Bosselaers IE, Scholl JH, Geertsma RE (2017) Nanomedicinal products: a survey on specific toxicity and side effects. Int J Nanomed 12:6107–6129
Cabanes A, Even-Chen S, Zimberoff J, Barenholz Y, Kedar E, Gabizon A (1999) Enhancement of antitumor activity of polyethylene glycol-coated liposomal doxorubicin with soluble and liposomal interleukin 2. Clin Cancer Res 5(3):687–693
Cagel M, Grotz E, Bernabeu E, Moretton MA, Chiappetta DA (2017) Doxorubicin: Nanotechnological overviews from bench to bedside. Drug Discov Today 22(2):270–281
Chan S, Davidson N, Juozaityte E, Erdkamp F, Pluzanska A, Azarnia N, Lee LW (2004) Phase III trial of liposomal doxorubicin and cyclophosphamide compared with epirubicin and cyclophosphamide as first-line therapy for metastatic breast cancer. Ann Oncol 15(10):1527–1534
Choi YP, Shim HS, Gao MQ, Kang S, Cho NH (2011) Molecular portraits of intratumoral heterogeneity in human ovarian cancer. Cancer Lett 307(1):62–71
Danhier F (2016) To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release 244(Pt A):108–121
Dimopoulos MA, Pouli A, Zervas K, Grigoraki V, Symeonidis A, Repoussis P, Mitsouli C, Papanastasiou C, Margaritis D, Tokmaktsis A, Katodritou I, Kokkini G, Terpos E, Vyniou N, Tzilianos M, Chatzivassili A, Kyrtsonis MC, Panayiotidis P, Maniatis A, Greek Myeloma Study Group (2003) Prospective randomized comparison of vincristine, doxorubicin and dexamethasone (VAD) administered as intravenous bolus injection and VAD with liposomal doxorubicin as first-line treatment in multiple myeloma. Ann Oncol 14(7):1039–1044
Dobrovolskaia MA, Germolec DR, Weaver JL (2009) Evaluation of nanoparticle immunotoxicity. Nat Nanotechnol 4(7):411–414
Dong J, Chen H (2018) Cardiotoxicity of anticancer therapeutics. Front Cardiovasc Med 5:9
Fang J, Nakamura H, Maeda H (2011) The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 63(3):136–151
Gabizon A, Tzemach D, Mak L, Bronstein M, Horowitz AT (2002) Dose dependency of pharmacokinetics and therapeutic efficacy of PEGylated liposomal doxorubicin (DOXIL®) in murine models. J Drug Target 10(7):539–548
Gabizon AA, Patil Y, La-Beck NM (2016) New insights and evolving role of PEGylated liposomal doxorubicin in cancer therapy. Drug Resist Updat 29:90–106
Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST (2017) Challenges and strategies in anti-cancer nanomedicine development: an industry perspective. Adv Drug Deliv Rev 108:25–38
Harris L, Batist G, Belt R, Rovira D, Navari R, Azarnia N, Welles L, Winer E, TLC D-99 Study Group (2002) Liposome-encapsulated doxorubicin compared with conventional doxorubicin in a randomized multicenter trial as first-line therapy of metastatic breast carcinoma. Cancer 94(1):25–36
Hong RL, Huang CJ, Tseng YL, Pang VF, Chen ST, Liu JJ, Chang FH (1999) Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C-26 tumor-bearing mice: Is surface coating with polyethylene glycol beneficial? Clin Cancer Res 5(11):3645–3652
Hunault-Berger M, Leguay T, Thomas X, Legrand O, Huguet F, Bonmati C, Escoffre-Barbe M, Legros L, Turlure P, Chevallier P, Larosa F, Garban F, Reman O, Rousselot P, Dhedin N, Delannoy A, Lafage-Pochitaloff M, Bene MC, Ifrah N, Dombret H, Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) (2011) A randomized study of PEGylated liposomal doxorubicin versus continuous-infusion doxorubicin in elderly patients with acute lymphoblastic leukemia: The GRAALL-SA1 study. Haematologica 96(2):245–252
Ilinskaya AN, Dobrovolskaia MA (2014) Immunosuppressive and anti-inflammatory properties of engineered nanomaterials. Br J Pharmacol 171(17):3988–4000
Jehn CF, Boulikas T, Kourvetaris A, Kofla G, Possinger K, Luftner D (2008) First safety and response results of a randomized phase III study with liposomal platin in the treatment of advanced squamous cell carcinoma of the head and neck (SCCHN). Anticancer Res 28(6B):3961–3964
Johnson KR, Ringland C, Stokes BJ, Anthony DM, Freemantle N, Irs A, Hill SR, Ward RL (2006) Response rate or time to progression as predictors of survival in trials of metastatic colorectal cancer or non-small-cell lung cancer: A meta-analysis. Lancet Oncol 7(9):741–746
Judson I, Radford JA, Harris M, Blay JY, van Hoesel Q, le Cesne A, van Oosterom AT, Clemons MJ, Kamby C, Hermans C, Whittaker J,, Verweij J, Nielsen S (2001) Donato di Paola E. Randomised phase II trial of PEGylated liposomal doxorubicin (DOXIL®/CAELYX) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: A study by the EORTC soft tissue and bone sarcoma group. Eur J Cancer 37(7):870–877
Keller AM, Mennel RG, Georgoulias VA, Nabholtz JM, Erazo A, Lluch A, Vogel CL, Kaufmann M, von Minckwitz G, Henderson IC, Mellars L, Alland L, Tendler C (2004) Randomized phase III trial of PEGylated liposomal doxorubicin versus vinorelbine or mitomycin C plus vinblastine in women with taxane-refractory advanced breast cancer. J Clin Oncol 22(19):3893–3901
Kosmas C, Angel J, Athanasiou A, Rapti A, Karanikas C, Lambaki S, Politis N, Mylonakis N (2009) 9088 phase III study of lipoplatin plus gemcitabine versus cisplatin plus gemcitabine in advanced NSCLC; interim analysis. Eur J Cancer Suppl 7(2):531
Latagliata R, Breccia M, Fazi P, Iacobelli S, Martinelli G, Di Raimondo F, Sborgia M, Fabbiano F, Pirrotta MT, Zaccaria A, Amadori S, Caramatti C, Falzetti F, Candoni A, Mattei D, Morselli M, Alimena G, Vignetti M, Baccarani M, Mandelli F (2008) Liposomal daunorubicin versus standard daunorubicin: Long term follow-up of the GIMEMA GSI 103 AMLE randomized trial in patients older than 60 years with acute myelogenous leukaemia. Br J Haematol 143(5):681–689
Maeda H, Tsukigawa K, Fang J (2016) A retrospective 30 years after discovery of the enhanced permeability and retention effect of solid tumors: Next-generation chemotherapeutics and photodynamic therapy–problems, solutions, and prospects. Microcirculation 23(3):173–182
Moghimi SM (2014) Cancer nanomedicine and the complement system activation paradigm: Anaphylaxis and tumour growth. J Control Release 190:556–562
Mylonakis N, Athanasiou A, Ziras N, Angel J, Rapti A, Lampaki S, Politis N, Karanikas C, Kosmas C (2010) Phase II study of liposomal cisplatin (lipoplatin) plus gemcitabine versus cisplatin plus gemcitabine as first line treatment in inoperable (stage IIIB/IV) non-small cell lung cancer. Lung Cancer 68(2):240–247
Nichols JW, Bae YH (2014) EPR: Evidence and fallacy. J Control Release 190:451–464
O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C, CAELYX Breast Cancer Study Group (2004) Reduced cardiotoxicity and comparable efficacy in a phase III trial of PEGylated liposomal doxorubicin HCl (CAELYX/Doxil®) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol 15(3):440–449
Olivieri J, Perna GP, Bocci C, Montevecchi C, Olivieri A, Leoni P, Gini G (2017) Modern management of anthracycline-induced cardiotoxicity in lymphoma patients: low occurrence of cardiotoxicity with comprehensive assessment and tailored substitution by nonPEGylated liposomal doxorubicin. Oncologist 22(4):422–431
Pan XQ, Zheng X, Shi G, Wang H, Ratnam M, Lee RJ (2002) Strategy for the treatment of acute myelogenous leukemia based on folate receptor beta-targeted liposomal doxorubicin combined with receptor induction using all-trans retinoic acid. Blood 100(2):594–602
Pastorino F, Brignole C, Marimpietri D, Sapra P, Moase EH, Allen TM, Ponzoni M (2003) Doxorubicin-loaded fab’ fragments of anti-disialoganglioside immunoliposomes selectively inhibit the growth and dissemination of human neuroblastoma in nude mice. Cancer Res 63(1):86–92
Pastorino F, Di Paolo D, Piccardi F, Nico B, Ribatti D, Daga A, Baio G, Neumaier CE, Brignole C, Loi M, Marimpietri D, Pagnan G, Cilli M, Lepekhin EA, Garde SV, Longhi R, Corti A, Allen TM, Wu JJ, Ponzoni M (2008) Enhanced antitumor efficacy of clinical-grade vasculature-targeted liposomal doxorubicin. Clin Cancer Res 14(22):7320–7329
Petersen GH, Alzghari SK, Chee W, Sankari SS, La-Beck NM (2016) Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin. J Control Release 232:255–264
Petschauer JS, Madden AJ, Kirschbrown WP, Song G, Zamboni WC (2015) The effects of nanoparticle drug loading on the pharmacokinetics of anticancer agents. Nanomedicine (Lond) 10(3):447–463
Rifkin RM, Gregory SA, Mohrbacher A, Hussein MA (2006) PEGylated liposomal doxorubicin, vincristine, and dexamethasone provide significant reduction in toxicity compared with doxorubicin, vincristine, and dexamethasone in patients with newly diagnosed multiple myeloma: A phase III multicenter randomized trial. Cancer 106(4):848–858
Roy AC, Park SR, Cunningham D, Kang YK, Chao Y, Chen LT, Rees C, Lim HY, Tabernero J, Ramos FJ, Kujundzic M, Cardic MB, Yeh CG, de Gramont A (2013) A randomized phase II study of PEP02 (MM-398), irinotecan or docetaxel as a second-line therapy in patients with locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma. Ann Oncol 24(6):1567–1573
Sabnani MK, Rajan R, Rowland B, Mavinkurve V, Wood LM, Gabizon AA, La-Beck NM (2015) Liposome promotion of tumor growth is associated with angiogenesis and inhibition of antitumor immune responses. Nanomedicine 11(2):259–262
Satalkar P, Elger BS, Hunziker P, Shaw D (2016) Challenges of clinical translation in nanomedicine: a qualitative study. Nanomedicine 12(4):893–900
Schell RF, Sidone BJ, Caron WP, Walsh MD, White TF, Zamboni BA, Ramanathan RK, Zamboni WC (2014) Meta-analysis of inter-patient pharmacokinetic variability of liposomal and non-liposomal anticancer agents. Nanomedicine 10(1):109–117
Smorenburg CH, de Groot SM, van Leeuwen-Stok AE, Hamaker ME, Wymenga AN, de Graaf H, de Jongh FE, Braun JJ, Los M, Maartense E, van Tinteren H, Nortier JW, Seynaeve C (2014) A randomized phase III study comparing PEGylated liposomal doxorubicin with capecitabine as first-line chemotherapy in elderly patients with metastatic breast cancer: results of the OMEGA study of the dutch breast cancer research group BOOG. Ann Oncol 25(3):599–605
Sparano JA, Makhson AN, Semiglazov VF, Tjulandin SA, Balashova OI, Bondarenko IN, Bogdanova NV, Manikhas GM, Oliynychenko GP, Chatikhine VA, Zhuang SH, Xiu L, Yuan Z, Rackoff WR (2009) PEGylated liposomal doxorubicin plus docetaxel significantly improves time to progression without additive cardiotoxicity compared with docetaxel monotherapy in patients with advanced breast cancer previously treated with neoadjuvant-adjuvant anthracycline therapy: Results from a randomized phase III study. J Clin Oncol 27(27):4522–4529
Stathopoulos GP, Antoniou D, Dimitroulis J, Michalopoulou P, Bastas A, Marosis K, Stathopoulos J, Provata A, Yiamboudakis P, Veldekis D, Lolis N, Georgatou N, Toubis M, Pappas C, Tsoukalas G (2010) Liposomal cisplatin combined with paclitaxel versus cisplatin and paclitaxel in non-small-cell lung cancer: A randomized phase III multicenter trial. Ann Oncol 21(11):2227–2232
Steimbach LM, Tonin FS, Virtuoso S, Borba HH, Sanches AC, Wiens A, Fernandez-Llimos F, Pontarolo R (2017) Efficacy and safety of amphotericin B lipid-based formulations-A systematic review and meta-analysis. Mycoses 60(3):146–154
Vici P, Pizzuti L, Gamucci T, Sergi D, Conti F, Zampa G, Del Medico P, De Vita R, Pozzi M, Botti C, Di Filippo S, Tomao F, Sperduti I, Di Lauro L (2014) Non-PEGylated liposomal doxorubicin-cyclophosphamide in sequential regimens with taxanes as neoadjuvant chemotherapy in breast cancer patients. J Cancer 5(6):398–405
von Roemeling C, Jiang W, Chan CK, Weissman IL, Kim BY (2017) Breaking down the barriers to precision cancer nanomedicine. Trends Biotechnol 35(2):159–171
Wu J, Lee A, Lu Y, Lee RJ (2007) Vascular targeting of doxorubicin using cationic liposomes. Int J Pharm 337(1–2):329–335
Xing M, Yan F, Yu S, Shen P (2015) Efficacy and cardiotoxicity of liposomal doxorubicin-based chemotherapy in advanced breast cancer: A meta-analysis of ten randomized controlled trials. PLoS One 10(7):e0133569
Yamaguchi N, Fujii T, Aoi S, Kozuch PS, Hortobagyi GN, Blum RH (2015) Comparison of cardiac events associated with liposomal doxorubicin, epirubicin and doxorubicin in breast cancer: a bayesian network meta-analysis. Eur J Cancer 51(16):2314–2320
Yang X, Zhang H, Nong J, Wang J, Li X, Zhang Q, Wang Q, Gao Y, Zhang S (2012) A randomized trial of liposomal paclitaxel plus cisplatin as first-line therapy for advanced non-small cell lung cancer. Zhongguo Fei Ai Za Zhi 15(4):208–212
Yardley DA, Burris HA, Spigel DR, Clark BL, Vazquez E, Shipley D, Barton J, Thompson D, Montes I, Greco FA, Hainsworth JD (2009) A phase II randomized crossover study of liposomal doxorubicin versus weekly docetaxel in the first-line treatment of women with metastatic breast cancer. Clin Breast Cancer 9(4):247–252
Yingchoncharoen P, Kalinowski DS, Richardson DR (2016) Lipid-based drug delivery systems in cancer therapy: What is available and what is yet to come. Pharmacol Rev 68(3):701–787
Acknowledgements
This work was supported by Woosuk University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author (M.K. Lee) declares that she has no conflict of interests.
Statement of human and animal rights
This article does not contain any studies with human or animal subjects performed by the author.
Rights and permissions
About this article

Cite this article
Lee, MK. Clinical usefulness of liposomal formulations in cancer therapy: lessons from the experiences of doxorubicin. J. Pharm. Investig. 49, 203–214 (2019). https://doi.org/10.1007/s40005-018-0398-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40005-018-0398-0