
Abstract
Background:
Carbonic anhydrase 1 (CA1) has been found to be involved in osteogenesis and osteoclast in various human diseases, but the molecular mechanisms are not completely understood. In this study, we aim to use siRNA and lentivirus to reduce or increase the expression of CA1 in Dental follicle stem cells (DFSCs), in order to further elucidate the role and mechanism of CA1 in osteogenesis, and provide better osteogenic growth factors and stem cell selection for the application of bone tissue engineering in alveolar bone fracture transplantation.
Methods:
The study used RNA interference and lentiviral vectors to manipulate the expression of the CA1 gene in DFSCs during in vitro osteogenic induction. The expression of osteogenic marker genes was evaluated and changes in CA1, alkaline phosphatase (ALP), Runt-related transcription factor 2 (RUNX2), and Bone morphogenetic proteins (BMP2) were measured using quantitative real-time polymerase chain reaction (qRT-PCR) and Western blotting (WB). The osteogenic effect was assessed through Alizarin Red staining.
Results:
The mRNA and protein expression levels of CA1, ALP, RUNX2, and BMP2 decreased distinctly in the si-CA1 group than other groups (p < 0.05). In the Lentivirus-CA1 (LV-CA1) group, the mRNA and protein expressions of CA1, ALP, RUNX2, and BMP2 were amplified to varying degrees than other groups (p < 0.05). Apart from CA1, BMP2 (43.01%) and ALP (36.69%) showed significant upregulation (p < 0.05). Alizarin red staining indicated that the LV-CA1 group produced more calcified nodules than other groups, with a higher optical density (p < 0.05), and the osteogenic effect was superior.
Conclusions:
CA1 can impact osteogenic differentiation via BMP related signaling pathways, positioning itself upstream in osteogenic signaling pathways, and closely linked to osteoblast calcification and ossification processes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Alveolar cleft is a complication of cleft lip with or without cleft palate (CL/P) and is usually treated by alveolar bone grafting [1]. However, both autologous bone graft [2], as the gold standard for bone regeneration, and various artificial bone graft materials including xenograft [3] and synthetic bone graft, resulting in limited practical applications due to their high incidence of side effects, limited donor resources and low osteogenic capacity [4]. Tissue engineering and regenerative medicine offer promising new treatments for this defect. In recent years, bone tissue engineering has been used to reconstruct bone tissue for alveolar cleft, but better scaffolds, stem cells and osteogenic growth factors that can promote its curative effect need to be explored [5].
The selection of stem cells is critical for the efficacy of tissue engineering [6]. Mesenchymal stem cells (MSCs) are widely regarded as one of the most suitable stem cell groups for bone regeneration, so they are used in bone tissue engineering [7, 8]. However, there are practical defects in clinical application, such as pain in collection method, donor site damage, and decreased proliferation and differentiation ability due to donor aging [9]. Thus, dental mesenchymal stem cells are emerging as a potential source of alternative cells which include dental pulp stem cells (DPSCs), periodontal ligament stem cells (PDLSCs), stem cells from human exfoliated deciduous teeth (SHEDs), dental follicular stem cells (DFSCs), etc. [10]. DFSCs are pluripotent stem cells derived from mesenchymal tissue and are true progenitor cells of periodontal tissue. Compared with other dental derived mesenchymal stem cells, DFSCs have stronger in vitro proliferation ability and higher expression level of osteogenic marker factors [11, 12]. The strong osteogenic ability makes DFSCs an attractive source of cells for repairing bone defects [13].
To achieve tissue regeneration, three critical elements are required: seed cells, scaffold materials, and cell growth factor [14]. To date, several studies have shown that osteogenic growth factors strongly affect various cell behaviors. For example, they can lead to self-renewal and more importantly, it plays an important role in stem cell differentiation [15, 16]. Even well-studied cell growth factors are subject to considerable controversy because studies based on different models often find different results and it is difficult to draw general conclusions. Therefore, the further study of osteogenic growth factors and their related genes is an important part of bone tissue engineering research [17].
As a member of the Carbonic Anhydrase (CA) family, CA1 plays a crucial role in catalyzing the reversible hydration and dehydration reaction of carbon dioxide and carbonic acid and promoting the formation of calcium carbonate [18, 19]. CA1 has been found to be involved in osteogenesis and osteoclast in various human diseases, including ankylosing spondylitis [20]. CA1 can promote joint calcification, ossification, and joint fusion by accelerating calcium carbonate deposition [21].
In view of the biological characteristics of CA1 in promoting calcification and ossification in vitro and the excellent osteogenic differentiation and proliferation ability of DFSCs, CA1 as an osteogenic inducer combined with DFSCs has a broad application prospect in bone tissue engineering. In combination with the research progress of alveolar cleft bone grafting and bone tissue engineering, the research used siRNA and lentivirus to reduce or increase the expression of CA1 in the osteogenic process of DFSCs, so as to further clarify the role and mechanism of CA1 in the osteogenic process. Ultimately, it provide better stem cell and osteogenic growth factor options for the application of bone tissue engineering in alveolar cleft bone grafting.
2 Material and methods
2.1 Cell culture and passage of DFSCs
This study was approved by the Ethics Committee of Qingdao University (Approval Number: QYFYWZLL27632), and all samples were obtained with informed consent from patients aged 18–25 years with good periodontal health who were admitted to the Affiliated Hospital of Qingdao University for orthodontic or impacted tooth extraction. All patients provided informed consent. In a biosafety cabinet, the follicle epithelium was preliminarily resected after repeated rinsing with phosphate buffered saline (PBS, Solarbio, Beijing, China) containing 10% penicillin–streptomycin mixture (Solarbio). The dental follicle tissue was immersed in a prepared digestive solution containing DispaseII enzyme (Solarbio, 4 mg/ml concentration) and collagenase type I (Solarbio, 3 mg/ml concentration) and digested for 50 min at 37 °C. Digestion was continued either in a 90 min 37 °C water bath or at 4 °C overnight. After digestion, medium was added to stop digestion, and the dental follicle tissue was centrifuged at 1000 rpm for 5 min. The dental follicle tissue was divided into more than ten small tissue blocks of about 1 mm2 using sterile blades. The above tissue blocks and culture medium were transferred to cell culture bottles, with about 1–3 tissues per bottle, and add 5 ml of alpha-MeM culture medium (BI, Kibbutz, Israel) containing 20% Fetal Bovine Serum (FBS BI) and 2% dual antibody (Solarbio). The bottles were placed in a cell incubator (37 °C/5% CO2, Thermo Corporation, Waltham, MA, USA) for culture. The culture medium was changed for the first time after 5 days and then every 3 days thereafter. When the cell fusion degree reached 70–80%, the cells were digested, and the cell density was diluted to 1 cell/200 μl after re-suspension. The cell suspension was transferred to 96-well plates for culture with 200 μl per well. When the cell confluence reached the standard of passable passage, passable passage could be performed.
2.2 Flow cytometry
DFSCs from generations 3–5 were digested with Trypsin digestion solution (0.25%, does not contain EDTA and phenol red, Solarbio), centrifuged, and suspended in 2 ml PBS. The cells were washed twice by centrifuging at 1500 rpm for 10 min and then suspended in pre-cooled PBS (containing 0.1% BSA). Strict cell counts were performed, and 1 × 106 DFSCs were diluted to 100 μl. Five 1.5 ml sterile EP tubes were taken, and 100 μl of cell suspension was added to each tube. Afterward, 5 μl of CD29-FITC, CD45-FITC, CD73-FITC, CD90-FITC flow cytometry antibodies (Elabscience, Wuhan, China) were added into the cell suspension prepared in the previous step. All samples were incubated for 30 min at 4 °C and protected from light. Following incubation, the cells were centrifuged at 1500 rpm for 10 min, and the supernatant was carefully discarded. The cell precipitate was re-suspended in 100 μl pre-cooled PBS, and the centrifugation procedure was repeated twice. The cells were then precipitated with 500 μl of cold PBS, stored at low temperature, protected from light, and tested on the A50 micro plus Flow Cytometry (Apogee, United Kingdom) as soon as possible. Finally, FlowJo V10 was used to analyze the flow cytometry data.
2.3 Alizarin red S staining
DFSCs were seeded into a 6-well plate at a density of 2 × 104 cells per well. Once the cell fusion degree reached 80%, the culture medium was replaced with a osteogenic induction culture medium containing 10% FBS, 0.15 μM dexamethasone, 10 μM β-Sodium glycerophosphate, and 10 μM ascorbic acid. The medium was changed every 3 days, and the cells were cultured for 28 days.
After 28 days, the culture medium was discarded and the cells were fixed at room temperature for 20–30 min. Next, the cells were washed three times with double steam water and then treated with alizarin red S dye (Procell, China)solution at room temperature for 10–20 min. The mineralized nodules that form was then observed under a microscope and photographed. To quantify the amount of mineralization, 10% cetylpyridine chloride (500 µl per well) was added to each well and the plate was incubated in a 37-degree incubator for 1 h. The absorbance of each well was then measured at 560 nm using an enzyme marker.
2.4 Oil red O staining
DFSCs were seeded into a 6-well plate at a density of 2 × 104 cells per well. Once the cell fusion degree reached 100%, the culture medium of the lipid formation group was replaced with a lipid induction medium containing 10% FBS, 1 μM dexamethasone, 200 μM indomethacin, 10 μM insulin, and 0.5 mM IBMX. The medium was changed every 3 days, and the cells were cultured for 14 days.
After 14 days, the culture medium was discarded, and the cells were fixed at room temperature for 20–30 min. The cells were then rinsed with double steaming water three times, and 60% isopropyl alcohol was added to soak the cells for 5 min. After the isopropyl alcohol was discarded, the cells were treated with oil red O working solution (Procell, China) and stained at room temperature for 20 min. Lipid droplets were then observed under a microscope and photographed.
2.5 Small interfering RNA (siRNA) transfection
Three groups of cells were set up: the si-CA1 group, the negative control group, and the blank control group. The si-CA1 group was transfected with GenOFFTM st-h-CA1 (RiboBio, China), the silenced siRNA of CA1, while the negative control group was transfected with a non-specific, negative control siRNA. The blank control group did not undergo transfection. Once the cells had reached optimal growth, they were digested with pancreatic enzymes and a cell suspension of 5 × 106 cells per well was prepared and inoculated into a 6-well plate. After 8–12 h, the culture medium was replaced with non-antibody complete medium, and 137 μl of freshly prepared transfection mixture was added drop by drop into each well and mixed gently. The plate was then placed back into the cell incubator. After 24 h of culture, the bone induction medium was changed and the cells were cultured for an additional 3 days. This medium contains specific factors that stimulate the cells to differentiate into osteoblasts and begin producing mineralized bone tissue.
2.6 Lentivirus transfection and osteogenic culture
To determine the optimal transfection conditions, three different groups of cells were established for the experiment. The cells in each group were transfected with either transfection reagent A, transfection reagent P, or no transfection reagent. Lentivirus (genechem, China) was diluted into three different concentrations: 1 × 108 PFU/mL, 5 × 107 PFU/mL, and 1 × 106 PFU/mL. Viruses of equal volume but different titers were added to the complex wells, resulting in MOI of 100, 50, and 10, respectively. Adherent P2–P5 cells were digested with trypsin and prepared in a 5 × 104 cells/mL cell suspension. The cells were then inoculated into a 96-well plate at a density of 100 μL/well, taking care to focus on the edge holes of the plate. Once the cell fusion degree reached 40–60%, the culture solution was aspirated, and the corresponding infection solution and virus solution were added according to the instructions. After 8–12 h, all cells were replaced with medium. During this period, careful attention was paid to the growth state of the cells, and the medium was changed in advance if the cell state was found to be altered. After forty eight hours of infection, the transfection was observed under an inverted fluorescence microscope. The infection efficiency was found to be between 80 and 90%, and the corresponding infection conditions of the group with good cell growth state were used for subsequent experiments.
The cells were divided into three groups in a six-hole plate: Lentivirus-CA1 (LV-CA1) group, negative control group, and blank control group. The LV-CA1 group was treated with complete medium + LV-CA1, the negative control group was treated with complete medium + negative control virus, and the blank control group was treated with complete medium only. Adherent P2–P5 cells were digested with pancreatic enzymes, and a 5 × 104 per well of cell suspension was prepared and inoculated into 6-well plates. When the cell fusion degree reached 40–60%, the culture solution was aspirated, and the corresponding infection solution and virus solution were added according to the instructions. Eight to twelve hours after transfection, all cells were changed to osteogenic induction medium. The growth state of cells was closely monitored during the process, and the medium was changed in advance if the cell state was found to be altered. Osteogenic induction medium was used throughout the 28-day osteogenic induction, with medium changes every 3 days. During the osteogenic culture, mRNA and protein of each group were extracted at 3 days 3, 7, 10, 14, 21, and 28, respectively, for detection.
2.7 Western blotting (WB)
Osteogenic medium was removed and adherent cells were scraped off and centrifuged. The supernatant was collected and protein concentration was determined with the BCA kit (Solarbio). The polyacrylamide gel electrophoresis stage involved the preparation of the glue, loading of the samples, and running of the gel. The transfer film stage involved transferring the separated proteins onto a PVDF membrane (Elabscience). The sealing stage involved incubating the membrane in a solution and blocking unspecific binding sites. The incubation of primary and secondary antibodies followed, and finally, the PVDF membrane was covered with ECL luminescent solution (Elabscience) and placed in a gel imager for image acquisition.
2.8 Quantitative real-time polymerase chain reaction (qRT-PCR)
The culture medium was removed and Trizol was added into each sample well. The lysate was collected and centrifuged before adding chloroform and isopropyl alcohol for precipitation. The RNA was measured for concentration and purity, and the reverse transcription reaction was carried out as per the kit instructions (Takara, Osaka, Japan). The Light Cycler 480 system (Roche, Indianapolis, IN, USA) was used for the PCR reaction, and the matching degree of the primers was determined by analyzing the amplification curve and dissolution curve of Real Time PCR. The details of the primers used are given in Table 1. Transcription conditions: 37 °C, 15 min; 85 °C, 5 s; Store at 4 °C. (If long-term storage is required, it should be stored at −20 °C or lower temperature).
2.9 Statistical analysis
All experiments were repeated three or more times. Statistics are expressed as mean and standard deviation (mean ± standard deviation) values. Statistical analysis was performed using SPSS 25.0 (Chicago, IL, USA) software and GraphPad Prism 8.0 (San Diego, CA, USA). The One-Way Analysis of Variance (ANOVA) test examined the statistical differences among the groups, and p < 0.05 was considered statistically significant.
3 Results
3.1 Identification and differentiation potential of DFSCs
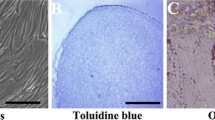
DFSCs were isolated from healthy dental follicles by enzyme digestion. After 7–10 days, the adherent growth of single cells or tissue blocks was observed. DFSCs were long spindle-shaped fibroblast-like cells with good activity, fast growth rate, and full cell bodies. After about 2 weeks of primary cell culture, the fusion rate reached 80%, and subculture could be repeated every 3 days on average. After 4 weeks of cell bone differentiation induction, mineralized nodules were observed and stained red by Alizarin red S dye solution. No red mineralized nodules were found in the non-induced group, indicating that the cells had the potential to differentiate into osteoblasts. After 14 days of lipid differentiation induction, circular lipid droplets were formed, which were stained orange by oil red O staining. No lipid droplets were observed in the non-induced group, indicating that the cells had the potential to differentiate into adipoid cells. Flow cytometry showed that DFSCs highly expressed CD29, CD73, and CD90, and were almost unexpressed in CD45 (Fig. 1). The cells isolated and cultured from the dental follicle were indeed DFSCs, in accordance with the characteristics of mesenchymal derived cells.

A The adhesion of DFSCs tissue blocks (×10) and the observation of cell morphology (×40). B DFSCs have the potential of osteogenic differentiation as revealed by staining with Alizarin Red (×40). C DFSCs have the potential of adipogenic differentiation, as revealed by staining with Oil Red O (×40). D Flow cytometric analysis that DFSCs exhibit high expression levels of CD29 (95.6%), CD73 (95.8%), and CD90 (96.1%), while CD45 is scarcely expressed (4.88%)
3.2 Suppressing CA1 reduced the expression of osteogenic marker gene
After transfection with siRNA, continue osteogenic culture for 7 days. Using WB and qRT-PCR techniques to detect the expression of osteogenic related genes. The results of WB experiment indicated that the protein expressions of alkaline phosphatase (ALP), runt-related transcription factor 2 (RUNX2) and bone morphogenetic proteins2 (BMP2) were also decreased when CA1 expression was decreased during in vitro osteogenic induction (p < 0.05). The results of qRT-PCR were consistent with WB, and the protein expressions of ALP, RUNX2 and BMP2 were also decreased when CA1 expression was decreased during in vitro osteogenic induction (p < 0.05) (Fig. 2).

A RNA knockdown of CA1 decreased the expression of osteogenic differentiation marker genes during osteogenic induction, as detected by Western blotting. B RNA knockdown of CA1 decreased the expression of osteogenic differentiation marker genes during osteogenic induction, as determined by qRT-PCR. C The transfection efficiency of the virus under different transfection conditions under fluorescence microscopy
3.3 Overexpression of CA1 can promote osteogenesis in vitro
As can be seen from the Fig. 2, compared with other conditions, transfection agent P can achieve the best transfection effect when MOI = 100. On days 3, 7, 10, 14, 21 and 28 of osteogenic induction culture, compared with negative control group and blank control group, the protein expressions of CA1, ALP, RUNX2 and BMP2 in LV-CA1 group were increased to varying degrees (p < 0.05) (Fig. 3). In addition to CA1, BMP2 (43.01%) and ALP (36.69%) were significantly increased (p < 0.05). The PCR results were consistent with WB (Fig. 4). The expression of RUNX2 fluctuated throughout the whole cycle, and increased in the experimental group compared with the control group except on day 10. At the 3rd, 7th, 10th, 14th, 21st and 28th days of osteogenic induction culture, the protein expression of CA1 in blank control group was higher at the beginning of induction, namely on the 3rd day, then decreased, gradually increased with time, and reached a peak at the late stage of osteogenic induction culture (p < 0.05). In the alizarin red staining experiment (Fig. 5), on day 7, 14, 21, 28 of the osteogenic culture, more calcified nodules were observed under the microscope in LV-CA1 group than in the negative control group and the blank control group, and the measurement results of OD value were consistent with the observation (p < 0.05).

A–F Overexpression of CA1 increased the expression of osteogenic marker genes during osteogenic induction on the 3, 7, 10, 14, 21, 28 day of culture, as detected by Western blotting

A–F The impact of CA1 overexpression on the mRNA expression of ALP, RUNX2, and BMP2 during the 3, 7, 10, 14, 21, 28 day of culture was detected by qRT-PCR G The protein relative expression levels of CA1 at various time points during osteogenic induction, as detected by Western blotting. H The mRNA relative expression levels of CA1 at various time points during osteogenic induction were detected by qRT-PCR

A–B The osteogenic effects of each group at various time points during the process of osteogenic induction were observed under a microscope (× 40) and with the naked eye after staining with Alizarin Red. C The relative level of mineralization (OD) of each group at various time points during the process of osteogenic induction were measured after staining with Alizarin Red
4 Discussion
Before bone tissue engineering was widely studied, it was generally accepted that autologous bone or other bone substitutes were used for bone grafting for alveolar cleft. Autologous bone grafting is a common source of bone for clinical bone grafting because it is the gold standard for bone regeneration, but its practical application is limited due to the high incidence of side effects and limited bone capacity at the donor site [2]. There are other bone tissue replacements, such as allografts and xenografts and synthetic bone grafts. However, their disadvantages include immune rejection and pathogen transmission [3]. The application of synthetic grafts is also limited due to their poor integration with native tissue and limited osteogenic and osteogenic ability, resulting in slow regeneration or graft failure [4]. In recent years, more and more research on alveolar cleft bone repair has focused on bone tissue engineering, combining alveolar cleft bone repair with different stem cells, scaffolds and cytokines in order to achieve better osteogenic effect [22, 23]. For example, Sun et al. established a rabbit alveolar cleft model and proved that bone collagen granules combined with human umbilical cord mesenchymal stem cells have the effect of repairing alveolar cleft bone defects [24]. Because different models and differences in the internal and external environment will affect the final osteogenic effect, future research directions will focus on the exploration and development of more appropriate stem cells, growth factors and scaffolds [22].
As a core component of bone tissue engineering, the selection of stem cells plays an important role in the ultimate repair effect. BMSCs represent a pluripotent group of cells that possess remarkable abilities for self-renewal and multi-lineage differentiation and have good regenerative potential in bone tissue engineering [25, 26]. To obtain sufficient bone marrow mesenchymal stem cells is a prerequisite for clinical application, which requires in vitro culture and expansion. However, in vitro culture of bone marrow mesenchymal stem cells includes several problems: prolonged time, high manufacturing costs, contamination risks, and requirements for good manufacturing practice facilities [27]. For clinical applications, dental stem cells are considered an attractive source of cells for bone tissue engineering due to their advantages such as ease of access and preservation, and multidirectional differentiation potential [28]. Compared with SHEDs and DPSCs, DFSCs showed higher expression levels of osteogenic markers, such as RUNX2 and ALP [29]. DFSCs are less mature than PDLSCs [13]. In addition, DFSCs can be obtained from impacted teeth, which are often discarded as medical waste during dental procedures. Their strong osteogenic ability makes them an attractive source of cells for repairing bone defects [15]. Studies have shown promising results in repairing critical dimensional defects in the skull of immune-compromised rats [30, 31]. In addition, dental stem cells are being explored as potential treatments for autoimmune and/or inflammatory diseases, given their powerful anti-inflammatory properties [32, 33]. Lucaciu et al. investigated the osteogenic potential of DFSCs in combination with titanium implants coated with various bioactive materials [29]. Their results showed that DFSCs could effectively promote bone regeneration on the surface of titanium implants. In our study, dental capsule stem cells were able to pass through the 7th generation and still had proliferative activity, and the selected cells of the 2nd to 5th generation had excellent osteogenic differentiation potential. Considering their status as true precursors of alveolar bone cells and their excellent in vitro proliferation and osteogenic capabilities, DFSCs offer an attractive option for bone tissue engineering.
In addition to suitable seed cells, bone tissue engineering also requires the formation of osteogenic growth factors stimulating the cells through signaling pathways to achieve good therapeutic results. Bone morphogenetic proteins (BMPs), fibroblast growth factors (FGFs), platelet-derived growth factors (PDGF), and vascular endothelial growth factors (VEGF), as osteogenic growth factors, have been shown to play osteogenic roles in bone tissue engineering [34]. However, due to the continuous dilution and degradation of osteogenic growth factors released by scaffolds after implantation, large doses of these proteins are required to prolong their therapeutic effect [35]. This can lead to adverse outcomes including inflammation, an increased risk of potentially malignant tumors, and ectopic bone formation in areas near where bone regeneration is needed [34]. Besides, Wang et al. proposed that there are many intermediate links from the extracellular signal to the final osteoblastic effect. The research on osteoblastic growth factors should be far from stopped, and additional ideal osteoblastic growth factors should be explored to reduce the cost [36]. As an emerging osteoblastic growth factor, CA1 plays a crucial role in both physiological and pathological activities of calcification and mineralization. Studies have shown that CA1 overexpression may significantly promote ossification and new bone formation [37]. CA1 not only promotes the hydration reaction of CO2, but also promotes the formation of calcium carbonate solid precipitator by assisting the combination of bicarbonate and calcium. Although the average calcium carbonate deposition in bone tissue was only 10% [20], in the study of Vuola et al., the bone formation of calcium carbonate implants was significantly higher than that of hydroxyapatite implants [38]. Ripamonti et al. also found that partially transformed hydroxyapatite/calcium carbonate structures induced spontaneous bone differentiation [39]. Another study found that CA1 in calcification of the aortic tissue of mice and humans high level expression of CA1 expression induced vascular smooth muscle cells calcification, and affect cell proliferation, apoptosis, migration and production of cytokines [40]. In our study, alizarin red staining showed that on the 7th, 14th, 21st and 28th days of osteogenic culture, more calcified nodules were formed in LV-CA1 group than in the control group, indicating that CA1 promoted osteogenesis of DFSCs in vitro and has the potential to be used as one of the osteogenic stimulants to improve the efficacy of bone tissue engineering in clinical bone repair.
Our study also showed that the expression of ALP, RUNX2, BMP2 and other osteogenic genes in vitro osteogenic differentiation of dentist stem cells decreased after the expression of CA1 was reduced by small interfering RNA. These results suggest that CA1 may regulate the osteogenic differentiation of dental follicular stem cells by regulating the expression of the above osteogenic genes and play a crucial role in the early stage of osteogenic differentiation. In addition, the presence of ALP and carbonic anhydrase cotransporters suggests that CA1 may not only influence ALP through gene regulation, but also increase or decrease the intracellular content of ALP during bone formation through cotransport [41]. ALP is one of the earliest genes involved in the calcification mechanism, triggering calcium deposition in bone cells. The regulatory mechanisms of ALP expression are very complex, and interweaving signaling pathway networks have only recently been proposed [42]. BMP/RUNX2/osteoblast specific transcription factor (Osterix, Osx1) network is an important regulatory pathway that regulates osteoblast differentiation, chondrogenesis, and ALP expression [17]. BMPs have been shown to regulate chondrocyte proliferation independently, and BMP2 are particularly important for chondrocyte proliferation and differentiation [43]. BMP2 is an important participant in postnatal bone homeostasis, and the osteogenic signal it provides is crucial for the innate repair ability of bone [44]. Short-term expression of BMP2 is a sufficient and necessary condition for irreversible induction of bone formation. BMP-2 activates several key signaling pathways for osteogenesis, cell survival, and apoptosis [45]. BMPs play a critical role in bone/cartilage progenitor cell differentiation and each subsequent stage of endochondral osteogenesis by regulating RUNX2 and Osx1 expression throughout bone development [46]. RUNX2 is abundant in mature osteoblasts and osteoblast lineage cells and is involved in early and late osteoblast differentiation and bone formation [47]. Relative expression of RUNX2 mRNA in osteoblast mesenchymal stem cells was up-regulated, down-regulated, and re-upregulated at day 14, 21, and 28, which was confirmed by our experiments [48]. In addition, during the preosteogenic phase, the expression of OSX1 downstream of RUNX2 is upregulated, causing preosteogenic cells to differentiate into immature osteoblasts and begin expressing osteogenic marker genes [49]. Our experimental results showed that at days 3, 7, 10, 14, 21 and 28 of osteogenic induction culture, the expressions of CA1, ALP, RUNX2 and BMP2 proteins in LV-CA1 group were significantly increased compared with negative control group and blank control group. Except the 3rd day, the expression of CA1 showed a gradually increasing trend in the whole cycle. Our results suggest that CA1 modulates osteogenesis through ALP, RUNX2, and BMP2 in bmp related osteogenic signaling pathways, and may play a role in early and late osteogenic differentiation.
In conclusion, these findings suggest that CA1 can impact osteogenic differentiation via BMP related signaling pathways, positioning itself upstream in osteogenic signaling pathways, and closely linked to osteoblast calcification and ossification processes. This study offers theoretical and experimental support for in vivo experiments and clinical applications of CA1, combined with DFSCs and bone tissue engineering, for alveolar cleft repair.
Data availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
Gaihre B, Uswatta S, Jayasuriya AC. Reconstruction of craniomaxillofacial bone defects using tissue-engineering strategies with injectable and non-injectable scaffolds. J Funct Biomater. 2017;8:49.
Proussaefs P, Lozada J. The use of intraorally harvested autogenous block grafts for vertical alveolar ridge augmentation: a human study. Int J Periodontic Restor Dent. 2005;25:351–63.
Eppley BL, Pietrzak WS, Blanton MW. Allograft and alloplastic bone substitutes: a review of science and technology for the craniomaxillofacial surgeon. J Craniofac Surg. 2005;16:981–9.
Frenken JW, Bouwman WF, Bravenboer N, Zijderveld SA, Schulten EA, Ten Bruggenkate CM. The use of straumann bone ceramic in a maxillary sinus floor elevation procedure: a clinical, radiological, histological and histomorphometric evaluation with a 6-month healing period. Clin Oral Implants Res. 2010;21:201–8.
Holly D, Klein M, Mazreku M, Zamborsky R, Polak S, Danisovic L, et al. Stem cells and their derivatives-implications for alveolar bone regeneration: a comprehensive review. Int J Mol Sci. 2021;22.
Anitua E, Troya M, Zalduendo M. Progress in the use of dental pulp stem cells in regenerative medicine. Cytotherapy. 2018;20:479–98.
Tatullo M, Codispoti B, Paduano F, Nuzzolese M, Makeeva I. Strategic tools in regenerative and translational dentistry. Int J Mol Sci. 2019;20.
Abdel Meguid E, Ke Y, Ji J, El-Hashash AHK. Stem cells applications in bone and tooth repair and regeneration: New insights, tools, and hopes. J Cell Physiol. 2018;233:1825–35.
Ling LE, Feng L, Liu HC, Wang DS, Shi ZP, Wang JC, et al. The effect of calcium phosphate composite scaffolds on the osteogenic differentiation of rabbit dental pulp stem cells. J Biomed Mater Res A. 2015;103:1732–45.
Zhang W, Yelick PC. Tooth repair and regeneration: potential of dental stem cells. Trends Mol Med. 2021;27:501–11.
Zhang J, Ding H, Liu X, Sheng Y, Liu X, Jiang C. Dental follicle stem cells: tissue engineering and immunomodulation. Stem Cells Dev. 2019;28:986–94.
Morsczeck C, Schmalz G. Transcriptomes and proteomes of dental follicle cells. J Dent Res. 2010;89:445–56.
Luan X, Ito Y, Dangaria S, Diekwisch TG. Dental follicle progenitor cell heterogeneity in the developing mouse periodontium. Stem Cells Dev. 2006;15:595–608.
Saito MT, Silverio KG, Casati MZ, Sallum EA, Nociti FH Jr. Tooth-derived stem cells: update and perspectives. World J Stem Cells. 2015;7:399–407.
Takahashi K, Ogura N, Aonuma H, Ito K, Ishigami D, Kamino Y, et al. Bone morphogenetic protein 6 stimulates mineralization in human dental follicle cells without dexamethasone. Arch Oral Biol. 2013;58:690–8.
Qi J, Yu T, Hu B, Wu H, Ouyang H. Current biomaterial-based bone tissue engineering and translational medicine. Int J Mol Sci. 2021;22.
Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol. 2016;12:203–21.
Supuran CT. Carbonic anhydrases–an overview. Curr Pharm Des. 2008;14:603–14.
Boron WF. Evaluating the role of carbonic anhydrases in the transport of HCO3−-related species. Biochim Biophys Acta. 2010;1804:410–21.
Chang X, Zheng Y, Yang Q, Wang L, Pan J, Xia Y, et al. Carbonic anhydrase I (CA1) is involved in the process of bone formation and is susceptible to ankylosing spondylitis. Arthr Res Ther. 2012;14:R176.
Zheng Y, Xu B, Zhao Y, Gu H, Li C, Wang Y, et al. CA1 contributes to microcalcification and tumourigenesis in breast cancer. BMC Cancer. 2015;15:679.
Toyota A, Shinagawa R, Mano M, Tokioka K, Suda N. Regeneration in experimental alveolar bone defect using human umbilical cord mesenchymal stem cells. Cell Transplant. 2021;30:963689720975391.
Bajestan MN, Rajan A, Edwards SP, Aronovich S, Cevidanes LHS, Polymeri A, et al. Stem cell therapy for reconstruction of alveolar cleft and trauma defects in adults: a randomized controlled, clinical trial. Clin Implant Dent Relat Res. 2017;19:793–801.
Sun XC, Wang H, Li JH, Zhang D, Yin LQ, Yan YF, et al. Repair of alveolar cleft bone defects by bone collagen particles combined with human umbilical cord mesenchymal stem cells in rabbit. Biomed Eng Online. 2020;19:62.
Liu J, Yu F, Sun Y, Jiang B, Zhang W, Yang J, et al. Concise reviews: characteristics and potential applications of human dental tissue-derived mesenchymal stem cells. Stem Cells. 2015;33:627–38.
Xu L, Liu Y, Sun Y, Wang B, Xiong Y, Lin W, et al. Tissue source determines the differentiation potentials of mesenchymal stem cells: a comparative study of human mesenchymal stem cells from bone marrow and adipose tissue. Stem Cell Res Ther. 2017;8:275.
Khojasteh A, Kheiri L, Motamedian SR, Nadjmi N. Regenerative medicine in the treatment of alveolar cleft defect: a systematic review of the literature. J Craniomaxillofac Surg. 2015;43:1608–13.
Bar JK, Lis-Nawara A, Grelewski PG. Dental Pulp stem cell-derived secretome and its regenerative potential. Int J Mol Sci. 2021;22:12018.
Lucaciu O, Soritau O, Gheban D, Ciuca DR, Virtic O, Vulpoi A, et al. Dental follicle stem cells in bone regeneration on titanium implants. BMC Biotechnol. 2015;15:114.
Guo W, Chen L, Gong K, Ding B, Duan Y, Jin Y. Heterogeneous dental follicle cells and the regeneration of complex periodontal tissues. Tissue Eng Part A. 2012;18:459–70.
Sun J, Li J, Li H, Yang H, Chen J, Yang B, et al. tBHQ Suppresses osteoclastic resorption in xenogeneic-treated dentin matrix-based scaffolds. Adv Healthc Mater. 2017. https://doi.org/10.1002/adhm.201700127.
Li Z, Jiang CM, An S, Cheng Q, Huang YF, Wang YT, et al. Immunomodulatory properties of dental tissue-derived mesenchymal stem cells. Oral Dis. 2014;20:25–34.
Yang C, Li X, Sun L, Guo W, Tian W. Potential of human dental stem cells in repairing the complete transection of rat spinal cord. J Neural Eng. 2017;14:026005.
Laird NZ, Acri TM, Tingle K, Salem AK. Gene- and RNAi-activated scaffolds for bone tissue engineering: current progress and future directions. Adv Drug Deliv Rev. 2021;174:613–27.
James AW, LaChaud G, Shen J, Asatrian G, Nguyen V, Zhang X, et al. A review of the clinical side effects of bone morphogenetic protein-2. Tissue Eng Part B Rev. 2016;22:284–97.
Wang T, Zhang X, Bikle DD. Osteogenic differentiation of periosteal cells during fracture healing. J Cell Physiol. 2017;232:913–21.
Yuan L, Wang M, Liu T, Lei Y, Miao Q, Li Q, et al. Carbonic anhydrase 1-mediated calcification is associated with atherosclerosis, and methazolamide alleviates its pathogenesis. Front Pharmacol. 2019;10:766.
Vuola J, Bhling T, Kinnunen J, Hirvensalo E, Asko-Seljavaara S. Natural coral as bone-defect-filling material. J Biomed Mater Res. 2000;51:117–22.
Ripamonti U, Crooks J, Khoali L, Roden L. The induction of bone formation by coral-derived calcium carbonate/hydroxyapatite constructs. Biomaterials. 2009;30:1428–39.
Siller AF, Whyte MP. Alkaline phosphatase: discovery and naming of our favorite enzyme. J Bone Miner Res. 2018;33:362–4.
Millan JL, Whyte MP. Alkaline phosphatase and hypophosphatasia. Calcif Tissue Int. 2016;98:398–416.
Kemoun P, Laurencin-Dalicieux S, Rue J, Farges JC, Gennero I, Conte-Auriol F, et al. Human dental follicle cells acquire cementoblast features under stimulation by BMP-2/-7 and enamel matrix derivatives (EMD) in vitro. Cell Tissue Res. 2007;329:283–94.
Bottini M, Mebarek S, Anderson KL, Strzelecka-Kiliszek A, Bozycki L, Simao AMS, et al. Matrix vesicles from chondrocytes and osteoblasts: their biogenesis, properties, functions and biomimetic models. Biochim Biophys Acta Gen Subj. 2018;1862:532–46.
Florencio-Silva R, Sasso GR, Sasso-Cerri E, Simoes MJ, Cerri PS. Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed Res Int. 2015;2015:421746.
Gamez B, Rodriguez-Carballo E, Ventura F. BMP signaling in telencephalic neural cell specification and maturation. Front Cell Neurosci. 2013;7:87.
Silverio KG, Davidson KC, James RG, Adams AM, Foster BL, Nociti Jr FH, et al. Wnt/beta-catenin pathway regulates bone morphogenetic protein (BMP2)-mediated differentiation of dental follicle cells. J Periodontal Res. 2012;47:309–19.
Pan K, Sun Q, Zhang J, Ge S, Li S, Zhao Y, et al. Multilineage differentiation of dental follicle cells and the roles of Runx2 over-expression in enhancing osteoblast/cementoblast-related gene expression in dental follicle cells. Cell Prolif. 2010;43:219–28.
Koo KT, Lee SW, Lee MH, Kim KH, Jung SH, Kang YG. Time-dependent expression of osteoblast marker genes in human primary cells cultured on microgrooved titanium substrata. Clin Oral Implants Res. 2014;25:714–22.
Rendl M, Polak L, Fuchs E. BMP signaling in dermal papilla cells is required for their hair follicle-inductive properties. Genes Dev. 2008;22:543–57.
Acknowledgements
Research was funded by grants from the National Natural Science Foundation of China (81070817) and Natural Science Foundation of Shandong Province (ZR2010HM054; ZR2015HM022). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Funding
National Natural Science Foundation of China, 81070817, Wen-lin Xiao,Natural Science Foundation of Shandong Province, ZR2010HM054, Wen-lin Xiao, ZR2015HM022, Wen-lin Xiao
Author information
Authors and Affiliations
Contributions
Jin-ze Zhao and Ying-Ying Ge: conducted the experiments, performed data collection and/or assembly; data analysis and interpretation; and wrote the manuscript. Cong Li: data collection and analysis. Ling-fa Xue, Yao-xiang Xu, Jin Yue and Wen-lin Xiao: developed the study conception and design; performed data analysis and interpretation; administrative and manuscript proof.All authorsread and approvedthe manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical statement
This study was approved by the Ethics Committee of Qingdao University (Approval Number: QYFYWZLL27632), and all samples were obtained with informed consent from patients.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, Jz., Ge, YY., Xue, Lf. et al. CA1 Modulates the Osteogenic Differentiation of Dental Follicle Stem Cells by Activating the BMP Signaling Pathway In Vitro. Tissue Eng Regen Med 21, 855–865 (2024). https://doi.org/10.1007/s13770-024-00642-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13770-024-00642-4