
Abstract
Background:
Hepatic fibrosis (HF) is a histopathological change in the process of long-term liver injury caused by cytokine secretion and internal environment disturbance, resulting in excessive liver repair and fiber scar. Nogo-B protein is widely distributed in peripheral tissues and organs and can regulate the migration of endothelial cells by activating TGF-β1 in vascular remodeling after injury. Nogo-B has been shown to promote organ fibrosis. This study was to determine the role of Nogo-B in HF.
Methods:
An HF model was built by intraperitoneal injections with 20% carbon tetrachloride. Localization of Nogo-B was detected by FISH. The interaction between Nogo-B and BACE1 was confirmed by Co-IP. Autophagy flux was analyzed using tandem mRFP-GFP-LC3 fluorescence microscopy, electron microscopy, and western blotting. Detection of serum AST and ALT and H&E staining were utilized to detect the degree of liver injury. The HF was evaluated by Masson trichromatic staining. RT-qPCR, western blotting, and immunofluorescence were employed to detect relevant indicators.
Results:
Reducing Nogo-B suppressed AST and ALT levels, the accumulation of collagen I and α-SMA, and expressions of pro-fibrotic genes in mouse liver. BACE1 was a potential downstream target of Nogo-B. Nogo-B was upregulated in TGF-β1-activated hepatic stellate cells (HSCs). Knocking down Nogo-B caused the downregulation of pro-fibrotic genes and inhibited viability of HSCs. Nogo-B knockdown prevented CCL4-induced fibrosis, accompanied by downregulation of extracellular matrix. Nogo-B inhibited HSC autophagy and increased lipid accumulation. BACE1 knockdown inhibited HSC autophagy and activation in LX-2 cells.
Conclusion:
Nogo-B knockdown prevents HF by directly inhibiting BACe1-mediated autophagy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Hepatic fibrosis (HF) is a histopathological change in the process of long-term liver injury caused by cytokine secretion and internal environment disturbance, resulting in excessive liver repair and fiber scar [1]. HF is characterized by chronic liver injury caused by a variety of toxic metabolites and the imbalance of repair mechanism, leading to hepatic stellate cell (HSC) activation and then excessive deposition of extracellular matrix (ECM) [2, 3]. Clinical treatment of HF remains at the etiological level, and there is still a lack of effective anti-fibrosis drugs [4, 5]. Without effective intervention, nodules form in the HF tissue, destroy liver function and structure, develop into cirrhosis, lead to liver failure, and even progress to liver cancer.
Nogo protein is a member of the reticular protein family, which is a transmembrane protein. The gene encoding the Nogo protein is located in human chromosome 2p16.1–2p16.3, and the Nogo mRNA produced is translated into three homologs Nogo-A/B/C by different promoters and splicing methods [6]. Nogo-B, also known as reticular 4B, is located primarily in the endoplasmic reticulum [7]. It is a spliced variant of Nogo-A and is expressed in most tissues [8]. Nogo-B protein is widely distributed in peripheral tissues and organs and can regulate the migration of endothelial cells by activating TGF-β1 in vascular remodeling after injury [9]. Nogo-B has been shown to promote organ fibrosis [10]. Abnormally high plasma Nogo-B levels are associated with cirrhosis and Child–Pugh score [11]. Nogo-B can regulate endothelial and smooth muscle cell responses after injury to various organs/tissues, including blood vessels [12], lungs [13], kidneys [14], 29, and liver [6]. Nogo-B can mediate vascular remodeling, cell migration and diffusion, epithelial-mesenchymal transition, and tumor angiogenesis [15]. It is clearly illustrated that a lack of Nogo-B can block fibrosis/cirrhosis and portal hypertension in HF [16].
Autophagy is a key pathway for damaged cellular organelles and protein degradation, providing energy for eukaryotic cells to maintain homeostasis [17]. Autophagy is a necessary condition for coping with and adapting to environmental changes [18]. Autophagy-related genes (Atgs) regulate autophagy maturation [19]. Autophagy inhibits HSC fibrosis by regulating the degradation of lipid droplets and p62 [20, 21] and activates HSC [22]. Therefore, HSC activation is closely related to autophagy.
Therefore, we hypothesize that Nogo-B-mediated autophagy and apoptosis in HSCs are the regulatory mechanisms by which this compound alleviates HF and involves autophagy pathways.
2 Materials and methods
2.1 CCl4-induced HF in mice
Twenty one-month-old male C57BL/6 mice were inhaled with CCl4 for 12 weeks. The mice were given drinking water containing phenobarbital (0.35 g/L) 3 days before CCl4 inhalation to worsen fibrosis/cirrhosis. The mice were placed in a gas chamber (60 × 40 × 20 cm) and inhaled CCl4 three times a week. In the first 3 weeks, the inhalation time of CCl4 was 1–2 min, and then increased to 3–5 min thereafter. Five–seven days before the experiment, the inhalation of CCl4 was stopped and the consumption of phenobarbital was also stopped. All animal studies were approved by the Ethics Committee of The Second Medical Center and National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital. The mice were euthanized by air embolization.
2.2 LX‐2 activation
LX‐2 (Fudan IBS Cell Center, Shanghai, China) were maintained in DMEM (Sigma-Aldrich, St. Louis, MO, USA) containing 10% FBS (Gibco; Thermo Fisher Scientific, Waltham, MA, USA) and activated by TGF-β1 at 1.25, 2.5, 5, and 10 nM, respectively.
2.3 Nogo-B and si‐BACE1 transfection
Nogo-B-KD, BACE1 siRNA (si-BACE1), and negative controls were transfected into LX-2 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Nogo-B-KD and Nogo-B-NC were treated in C57BL/6 mice 4 times per week for 2 weeks using siRNA‐Mate™ transfection reagent (Gema, Shanghai, China).
2.4 HE and Masson staining
Fresh liver tissue (about 0.5 cm2) was fixed with 4% paraformaldehyde, embedded in paraffin, and prepared to 4 μm. H&E and Masson staining were performed to assess changes in liver damage and HF. Images were obtained under a BX-51 microscope (Olympus, Tokyo, Japan). The degree of HF was graded according to the metavir score, and each case was semi-quantitatively classified into 0–4 scores. 0 score indicated no fibrosis, 4 score indicated cirrhosis, and 1–3 score indicated portal vein enlargement with different degrees of diaphragm. HF was quantified and analyzed using ImageJ V1.8.0 software.
2.5 Serum biochemistry
Serum ALT and AST were measured by biochemical assays (C010-2-1 and C009-2-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Optical density was read at 510 nm.
2.6 RT-qPCR
Total RNA from LX-2 cells and liver tissue was obtained using RNAiso Plus reagent (9109; Takara; Beijing, China). cDNA was produced with Prime Script RT kit (RR036A; Takara). Gene quantification was conducted with the TB Green fluorescence quantitative PCR kit (RR420A; Takara) using Light Cycler 480 software. PCR procedures included 45 cycles of denaturation at 95 °C for 15 s and annealing at 60 °C for 30 s, followed by extension at 95 °C for 5 s. Target gene levels were analyzed using the 2−ΔΔCt method. Sequence primers are found in Table 1
2.7 Western blotting analysis
Total protein was obtained from LX-2 cells and mouse liver tissues by ice-incubation with a lysis buffer containing 1% PMSF and RIPA and centrifuged at 4 °C at 12,500 rpm for 15 min. The supernatant quantified with a BCA kit (P1511-2; Applygen; Beijing, China) was mixed with SDS protein loading buffer, denatured at 100 °C for 10 mi, separated by 10% SDS–polyacrylamide gel (P0012A; Beyotime, Haimen, China), immediately transferred to the PVDF membrane (HVLP02500; Millipore, Burlington, MA, USA), and incubated with primary antibodies at 4 °C overnight. After corresponding secondary antibody (1:2000) reaction 1 h, the protein membrane was visualized using ECL kits (ANT044; Antgene; Wuhan, China).
2.8 Cell proliferation assay
LX-2 cells (1 × 104 cells/ml) were cultured in the 96-well plate for 24 h and treated with TGF‐β1 (1.25, 2.5, 5, and 10 nM) for 24 h. After 10 μL CCK-8 reagent was incubated for 2 h, the absorbance at 450 nm was measured.
2.9 Immunofluorescence
Paraffin-embedded liver sections (4 μm) were immersed in xylene, dewaxed in graded ethanol, and then in hot citrate buffer (pH 6.0). Next, sections were treated with 0.3% Triton X-100, blocked with 1% donkey serum, and incubated overnight with LC3B (Sigma, 1:100) and α-smooth muscle actin (SMA) (Sigma, 1:100) at 4 °C. Alexa Fluor 488- and 594-coupled secondary antibodies (1:500) were supplemented for 1 h, followed by 1 μg/ml DAPI (C1002; Beyotime) for 5 min. Images were observed under a confocal laser scanning microscope (Olympus, FV-500, Tokyo, Japan).
2.9.1 Co-IP assay
Cells were prepared into a lysate, centrifuged at 12,000 rpm at 4 °C for 15 min, incubated with antibody or control IgG of interest at 4 °C overnight, and added with 10 μL Protein A + G agarose beads (78,610, Thermo Scientific) at 4 °C for 2 h. Finally, the sample was loaded into SDS-PAGE gel for Western blotting.
2.9.2 Autophagy flux analysis
LX-2 cells were transfected with mRFP-GFP-tandem fluorescently labeled LC3 using Lipofectamine 2000. GFP and mRFP were observed with Olympus FV1000 microscope (Olympus). The image was captured using FV10‐ASW3.0 software. Yellow dots (GFP signals combined with RFP signals) represent early autophagosomes, and red dots (RFP signals alone) represent late autophagosomes. Autophagy flux was assessed by color changes in GFP/mRFP.
2.9.3 FISH
FISH-based Nogo-B imaging was performed according to the previously described protocol [23] and a 5 'biotin-labeled probe for Nogo-B (CTGCCTGTCAAGGATGTTTACA) was obtained from Gema.
2.9.4 Statistical analysis
Data were expressed as mean ± SE and assessed by analysis of variance and student t-test. P < 0.05 was considered statistically significant.
3 Results
3.1 TGF‐β1 triggers activation and proliferation of LX‐2 cells
LX-2 cells were activated by TGF-β1 (1–10 ng/ml) and tested by CCK-8 assay. As shown, LX-2 cell proliferation was significantly enhanced by TGF-β1 at 1–5 ng/ml (Fig. 1A). However, TGF-β1 at 10 ng/ml, suppressed LX-2 cell proliferation (Fig. 1A), possibly because high doses of TGF-β1 stimulation led to excessive cytotoxic effects that promoted more apoptosis. α-SMA and Collagen I expressions were elevated in LX-2 cells treated with TGF-β1 at 10 ng/ml (Fig. 1B, C). Therefore, 10 ng/ml TGF-β1 was used for follow-up experiments.

TGF‐β1 induces activation and proliferation of LX‐2 cells. A CCK8 assay analyzed cell proliferation; B, C Western blot and RT-qPCR analysis of α-SMA and Collagen I
3.2 Nogo-B is upregulated in TGF-β1-activated HSCs
TGF-β1-induced LX-2 activation led to up-regulation of Nogo-B (Fig. 2A). Nogo-B level in LX-2 increased in response to TGF-β1 (Fig. 2B). FISH data showed a major increase in Nogo-B expression in the cytoplasm of TGF-β1-activated LX-2 cells (Fig. 2C).

Nogo-B is upregulated in TGF-β1-induced LX-2 cells. A, B Western blot and RT-qPCR analysis of Nogo-B; C FISH detection of Nogo-B in LX-2 cells
3.3 Knocking down Nogo-B inhibits LX-2 cell proliferation, activation, and ECM accumulation
TGF-β1-activated LX-2 cells were treated with Nogo-B knockdown, and LX-2 cell proliferation was found to decrease after Nogo-B knockdown (Fig. 3A). α-SMA and Collagen I protein expression was reduced (Fig. 3B), and α-SMA, Collagen I, and CTGF mRNA expressions were decreased (Fig. 3C–E). Immunofluorescence showed a significant reduction in α-SMA (Fig. 3F). Meanwhile, levels of TIMP-1 and CTGF decreased in LX-2 cells after Nogo-B knockdown (Fig. 3G). Decreased α-SMA, Collagen I, and Vimentin protein expression further supported the antifibrotic activity of Nogo-B knockdown in vitro (Fig. 3H).

Knocking down Nogo-B inhibits LX-2 cell proliferation, activation, and ECM production. A CCK8 assay analyzed cell proliferation; B Western blot analysis of α-SMA and Collagen I; C–E: RT-qPCR analysis of α-SMA, Collagen I, and CTGF; F Immunofluorescence detection of α-SMA expression; G–H: Western blot analysis of CTGF, TIMP-1, α-SMA, Collagen I, and Vimentin
3.4 Knocking down Nogo-B alleviates CCL4-induced liver injury and HF in mice
ALT and AST were reduced in CCL4-induced mice with Nogo-B knockdown (Fig. 4A, B). HE staining found that liver sections in CCl4-induced mice showed extensive proliferative fibrous tissue in portal triplet and a large number of hepatocyte edema/necrosis around the central vein, disappearance of hepatic cord, and formation of pseudo-lobe. Knocking down Nogo-B effectively relieved the destruction of liver tissue structure and reduced the proliferation of collagen fibers (Fig. 4C, D). Fibrosis score was correspondingly lower as assessed by pathophysiology (Fig. 4E).

Knocking down Nogo-B alleviates CCL4-induced liver injury and HF in mice. A, B Changes of serum ALT and AST concentrations; C HE-stained liver sections; D Masson-stained liver sections; E HF score
3.5 Knocking down Nogo-B alleviates CCL4-induced HF in mice by regulating autophagy
Western blotting and RT-qPCR results showed that CCl4 intervention led to LC3-II and Beclin1 accumulation, but no significant change in LC3-I, so the LC3-II/LC3-I ratio increased, while knocking down Nogo-B reversed LC3 and Beclin1 expression (Fig. 5A, B). Similarly, ULK1, ATG5/7/9, and Beclin1 mRNA levels were reduced by knocking down Nogo-B (Fig. 5C, D). p62 expression decreased with the intervention of CCl4, while increased after Nogo-B knockdown treatment (Fig. 5A, B). CCl4 intervention led to LC3-II accumulation and increased LC3-II/LC3-I ratio. Knocking down Nogo-B reversed LC3 and p62 expression (Fig. 5E–G).

Knocking down Nogo-B alleviates CCL4-induced HF in mice by regulating autophagy. A Western blot analysis of LC3, p62, and Beclin1; B RT-qPCR analysis of LC3, p62, and Beclin1; C RT-qPCR analysis of ULK1 and Beclin1; D RT-qPCR analysis of ATG5/7/9; E–G Immunofluorescence detection of LC3 and P62
3.6 TGF‐β1 induces autophagy and activation of LX-2 cells
The effect of Nogo-B knockdown on autophagy of HSCs in vitro was determined. Western blotting determined that with the increase of TGF‐β1 concentration, LC3-II/LC3-I and Beclin1 expression gradually increased, while p62 level gradually decreased (Fig. 6A, B). ULK1, ATG5/7/9, and Beclin1 mRNA expression also increased with the increase of TGF‐β1 concentration (Fig. 6C, D). In short, TGF‐β1 can stimulate the activation of LX-2 cells and induce autophagy.

TGF‐β1 induces autophagy of LX-2 cells. A Western blot analysis of LC3, p62, and Beclin1; B RT-qPCR analysis of LC3, p62, and Beclin1; C RT-qPCR analysis of ULK1 and Beclin1; D RT-qPCR analysis of ATG5/7/9
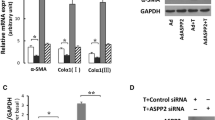
3.7 Down-regulation of BACE1 promotes the antifibrotic effect of Nogo-B knockdown
To explore the specific mechanism of Nogo-B knockdown to improve HF, BACE1 was identified as a potential downstream target of Nogo-B in protein–protein interaction networks (Fig. 7A). The interaction between Nogo-B and BACE1 was confirmed by Co-IP assay (Fig. 7B). BACE1 is the main neuronal β-secreting enzyme produced by amyloid-β, which is degraded in lysosomes and can maintain neuronal homeostasis in the autophagolysosomal system [24]. BACE1 is also involved in the regulation of autophagy [25]. After intervening BACE1 in mice, the degree of HF was assessed by histopathological analysis. The results showed that downregulating BACE1 alleviated histopathological changes in the liver of mice and could further improve the effect of Nogo-B knockdown. In addition, both HE and Masson staining showed that Nogo-B knockdown in combination with BACE1 downregulation improved fibrosis, manifested by reduced collagen deposition, reduced fibrotic area, and reduced inflammatory cell infiltration (Fig. 7C, D).

Down-regulating BACE1 promotes the antifibrotic effect of Nogo-B knockdown. A Protein interaction network analysis of Nogo-B; B Co-IP analysis of the interaction between Nogo-B and BACE1; C HE-stained liver sections; D Masson-stained liver sections
3.8 BACE1 knockdown can improve HSC autophagy and activation
The knockdown efficiency of BACE1 was detected by RT-qPCR and Western blotting in LX-2 cells (Fig. 8A, B). BACE1 knockdown significantly down-regulated α-SMA, TIMP-1, and Collagen I (Fig. 8C), suggesting that BACE1 knockdown inhibited HSCs activation. BACE1 knockdown in LX-2 cells also reduced the number of autophagosomes (Fig. 8D). Scanning electron microscope observed that the number of autophagy vacuoles in LX-2 cells was also reduced after downregulating BACE1 (Fig. 8E).

BACE1 knockdown can improve the autophagy and activation of HSCs. A, B Western blot and RT-qPCR analysis of BACE1 knockdown; C RT-qPCR analysis of α-SMA, Collagen I, and TIMP-1; D Immunofluorescence detection of autophagy flux; E Scanning electron microscopy observed autophagy vacuoles in LX-2 cells
4 Discussion
HF is an auto-defense reaction of the body after chronic liver damage. Repeated wound healing leads to HF, and its main histopathological changes are the excessive deposition of ECM in the liver [26]. It has been evidenced that antiviral treatment for chronic hepatitis B and C could slow down or stop HF progression [27]. Nogo-B silencing has been shown to prevent and reverse HF [28].
Our in vitro experiments showed that Nogo-B was highly expressed in TGF-β1-activated HSCs, and was mainly up-regulated in the cytoplasm. It has been reported that Nogo-B promotes congenital inflammation and contributes to hepatic ischemia and reperfusion injury [6]. Also, Nogo-B enhances angiogenesis and cardiac repair after myocardial infarction [29]. Our in vivo experiments also showed that Nogo-B was significantly upregulated in mouse liver fibrotic tissue. Nogo-B knockdown inhibited α-SMA, TIMP-1, and Collagen I and led to growth arrest of HSCs, suggesting that Nogo-B knockdown may target the matrix synthesis process by inhibiting HSC activation and proliferation, thereby negatively regulating HF. In the mouse model, Nogo-B knockdown significantly reduced the severity of HF, which can be achieved through collagen deposition and reduction in collagen content. In addition, Nogo-B knockdown has a liver protective effect on the process of HF by reducing AST and ALT and suppressing HSC activation.
Autophagy is an evolutionally conserved process of self-digestion in which cytoplasmic material is sequestered in cytoplasmic autophagosomes and delivered to lysosomes for degradation. As far as we know, Nogo-B is a gene involved in fibrosis and autophagy [16]. This study proved that Nogo-B may block HSC activation by inhibiting autophagy in HSCs.
BACE1, as an aspartate protease, belongs to the pepsin family [30] and was first discovered in 1999. It is especially abundant in various neuronal cell types and exerts in maintaining and repairing the central nervous system and peripheral nervous system, especially in response to injury and inflammation [31]. Nogo-B knockdown reduced BACE1 levels in HSCs, and BACE1 was down-regulated in the fibrotic animal model, suggesting that Nogo-B could regulate BACE1 expression. The results indicated that Nogo-B regulates HF by interacting with the BACE1 pathway, highlighting the therapeutic potential and potential mechanisms by which Nogo-B inhibits the BACE1 pathway for the prevention and treatment of HF.
This study originally explored the relationship between Nogo-B, autophagy, and HF and clarified that Nogo-B is upregulated in TGF-β1-activated LX-2 cells and mouse liver fibrotic tissues. Nogo-B has therapeutic potential in HF by inhibiting HSC activation, stimulating matrix degradation, and inhibiting collagen production. The molecular basis of Nogo-B's antifibrotic effect is the direct inhibition of BACE1 leading to HSC autophagy inhibition. These findings suggest that knockdown of Nogo-B may block HF through an autophagy-dependent pathway and therefore represent a new and promising therapeutic target for HF.
Data availability
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
References
Friedman SL, Pinzani M. Hepatic fibrosis 2022: unmet needs and a blueprint for the future. Hepatology. 2022;75:473–88.
Matsuda M, Seki E. Hepatic stellate cell-macrophage crosstalk in liver fibrosis and carcinogenesis. Semin Liver Dis. 2020;40:307–20.
Liu Yi, Que KT, Yang PF. TREM2 promotes liver fibrosis by enhancing hepatic stellate cell endocytosis and activation via phosphorylating ITGAV. J Biol Regul Homeost Agents. 2023;37:3693–704.
Khanam A, Saleeb PG, Kottilil S. Pathophysiology and treatment options for hepatic fibrosis: can it be completely cured? Cells. 2021;10:1097.
Parola M, Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. 2019;65:37–55.
Rao J, Cheng F, Zhou H, Yang W, Qiu J, Yang C, et al. Nogo-B is a key mediator of hepatic ischemia and reperfusion injury. Redox Biol. 2020;37:101745.
Oertle T, Schwab ME. Nogo and its paRTNers. Trends Cell Biol. 2003;13:187–94.
Teng FY, Tang BL. Cell autonomous function of Nogo and reticulons: The emerging story at the endoplasmic reticulum. J Cell Physiol. 2008;216:303–8.
Zhang Y, Huo W, Wen Y, Li H. Silencing Nogo-B receptor inhibits penile corpus cavernosum vascular smooth muscle cell apoptosis of rats with diabetic erectile dysfunction by down-regulating ICAM-1. PLoS One. 2019;14:e0220715.
Dong C, Liu Y, Jiang K, Wang H, Qu W, Zhang C, et al. The Nogo-B receptor promotes human hepatocellular carcinoma cell growth via the Akt signal pathway. J Cell Biochem. 2018;119:7738–46.
Wen M, Men R, Yang Z, Dan X, Wu W, Liu X, et al. The value of circulating Nogo-B for evaluating hepatic functional reserve in patients with cirrhosis. Dis Markers. 2015;2015:419124.
Yang Q, Zhang C, Xie H, Tang L, Liu D, Qiu Q, et al. Silencing Nogo-B improves the integrity of blood-retinal barrier in diabetic retinopathy via regulating Src, PI3K/Akt and ERK pathways. Biochem Biophys Res Commun. 2021;581:96–102.
Tadokoro KS, Rana U, Jing X, Konduri GG, Miao QR, Teng RJ. Nogo-B receptor modulates pulmonary artery smooth muscle cell function in developing lungs. Am J Respir Cell Mol Biol. 2016;54:892–900.
Hernandez-Diaz I, Pan J, Ricciardi CA, Bai X, Ke J, White KE, et al. Overexpression of circulating soluble Nogo-B improves diabetic kidney disease by protecting the vasculature. Diabetes. 2019;68:1841–52.
Li YK, Xie YJ, Wu DC, Long SL, Tang S, Mo ZC. Nogo-B receptor in relevant carcinoma: Current achievements, challenges and aims (Review). Int J Oncol. 2018;53:1827–35.
Zhao D, Xue C, Yang Y, Li J, Wang X, Chen Y, et al. Lack of Nogo-B expression ameliorates PPARγ deficiency-aggravated liver fibrosis by regulating TLR4-NF-κB-TNF-α axis and macrophage polarization. Biomed Pharmacother. 2022;153:113444.
Mallat A, Lodder J, Teixeira-Clerc F, Moreau R, Codogno P, Lotersztajn S. Autophagy: a multifaceted partner in liver fibrosis. Biomed Res Int. 2014;2014:869390.
Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16:661–75.
Antonioli M, Di Rienzo M, Piacentini M, Fimia GM. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. 2017;42:28–41.
Ba L, Gao J, Chen Y, Qi H, Dong C, Pan H, et al. Allicin attenuates pathological cardiac hypertrophy by inhibiting autophagy via activation of PI3K/Akt/mTOR and MAPK/ERK/mTOR signaling pathways. Phytomedicine. 2019;58:152765.
Varshney P, Saini N. PI3K/AKT/mTOR activation and autophagy inhibition plays a key role in increased cholesterol during IL-17A mediated inflammatory response in psoriasis. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1795–803.
Filali-Mouncef Y, Hunter C, Roccio F, Zagkou S, Dupont N, Primard C, et al. The ménage à trois of autophagy, lipid droplets and liver disease. Autophagy. 2022;18:50–72.
Xu J, Wu ZS, Wang Z, Le J, Zheng T, Jia L. Autonomous assembly of ordered metastable DNA nanoarchitecture and in situ visualizing of intracellular microRNAs. Biomaterials. 2017;120:57–65.
Feng T, Tammineni P, Agrawal C, Jeong YY, Cai Q. Autophagy-mediated regulation of BACE1 protein trafficking and degradation. J Biol Chem. 2017;292:1679–90.
Du X, Huo X, Yang Y, Hu Z, Botchway BOA, Jiang Y, et al. miR-124 downregulates BACE 1 and alters autophagy in APP/PS1 transgenic mice. Toxicol Lett. 2017;280:195–205.
Adori M, Bhat S, Gramignoli R, Valladolid-Acebes I, Bengtsson T, Uhlèn M, et al. Hepatic innervations and nonalcoholic fatty liver disease. Semin Liver Dis. 2023;43:149–62.
Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50:1294–306.
Tashiro K, Satoh A, Utsumi T, Chung C, Iwakiri Y. Absence of Nogo-B (reticulon 4B) facilitates hepatic stellate cell apoptosis and diminishes hepatic fibrosis in mice. Am J Pathol. 2013;182:786–95.
Zheng Y, Lin J, Liu D, Wan G, Gu X, Ma J. Nogo-B promotes angiogenesis and improves cardiac repair after myocardial infarction via activating Notch1 signaling. Cell Death Dis. 2022;13:306.
Cervellati C, Valacchi G, Zuliani G. BACE1: from biomarker to Alzheimer’s disease therapeutical target. Aging. 2021;13:12299–300.
Bazzari FH, Bazzari AH. BACE1 inhibitors for Alzheimer’s disease: the past, present and any future? Molecules. 2022;27:8823.
Acknowledgements
Not applicable.
Funding
National Natural Science Foundation of China (No. 81570563).
Author information
Authors and Affiliations
Contributions
LG designed the research study. YingJie Zhuang and ZhengYi Liu performed the research. LiLi Gao and ZhengYi Liu provided help and advice. LiLi Gao and YingJie Zhuang analyzed the data. LiLi Gao wrote the manuscript. LiLi Gao reviewed and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Ethical approval
The animal experiment research protocol was approved by the Ethics Committee of The Second Medical Center and National Clinical Research Center for Geriatric Diseases, Chinese PLA General Hospital (202002BJ63) and performed in accordance with the “Guidelines for the care and use of experimental animals.”
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gao, L., Zhuang, Y. & Liu, Z. Reducing Nogo-B Improves Hepatic Fibrosis by Inhibiting BACe1-Mediated Autophagy. Tissue Eng Regen Med 21, 777–789 (2024). https://doi.org/10.1007/s13770-024-00641-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13770-024-00641-5