
Abstract
Background:
Ever-growing tissue regeneration causes pressing need for large population of stem cells. However, extensive cell expansion eventually leads to impaired regenerative potentials. In this study, chromobox protein homolog 7 (CBX7) was overexpressed to rejuvenate late passage dental pulp stem cells (DPSCs-P9).
Methods:
The recruitment of copper ions (Cu2+)-activated hypoxia-inducible factor-1α (HIF-1α) to the CBX7 gene promoter was confirmed by chromatin immunoprecipitation assay. Functions subsequent to Cu2+-induced or recombinant overexpression of CBX7 on proliferation, multipotency, odontoblastic differentiation and angiogenesis were investigated in vitro, while murine subcutaneous transplantation model was used to further detect the effects of Cu2+-induced CBX7 overexpression in vivo.
Results:
Our data displayed that CBX7 overexpression maintain proliferation and multipotency of DPSCs-P9 almost as strong as those of DPSCs-P3. Both gene level of odontoblast-lineage markers and calcium precipitation were nearly the same between CBX7 overexpressed DPSCs-P9 and normal DPSCs-P3. Moreover, we also found upregulated expression of vascular endothelial growth factor in DPSCs-P9 with CBX7 overexpression, which increased the number of capillary-like structures and migrating co-cultured human umbilical vein endothelial cells as well. These findings indicate CBX7 as an effective factor to rejuvenate late passage stem cells insusceptible to cell expansion. Cu2+ has been proved to achieve CBX7 overexpression in DPSCs through the initiation of HIF-1α-CBX7 cascade. Under Cu2+ stimulation since P3, DPSCs-P9 exhibited ameliorated regenerative potential both in vitro and in vivo.
Conclusion:
Long-term stimulation of Cu2+ to overexpress CBX7 could be a new strategy to manufacture large population of self-renewing stem cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Stem cells are an attractive candidate for use in regenerative medicine because of their self-renewal and multipotency [1], as well as the paracrine effects [2]. However, extensive in vitro cell expansion eventually impairs these properties [3]. So, the loss of stem cell functions is a major factor restraining the clinical application of stem cells. Dental pulp stem cells (DPSCs), isolated from pulp tissue, possess high multipotent differentiation [4] and induce angiogenesis partially by paracrine secretion of abundant pro-angiogenic factors [5]. Overall, DPSCs are more suitable for mineralized tissue regeneration [6]. They differentiate into bone-like tissues when loaded on scaffolds in animal models [7], and regenerate bone in human grafts [8, 9]. Their aggregates auto-transplanted into injured immature permanent incisors achieve three-dimensional pulp tissue regeneration [10]. Due to the limited number of cells harvested from tiny pulp tissue (~ 10–100 μl), DPSCs need to be extended over multiple passages to obtain sufficient cells for therapeutic use but with inevitable decline of stemness and paracrine ability. Hence, strategies need to be proposed to resolve this problem, such as adding bioactive agents to cell expansion medium.
Chromobox protein homolog 7 (CBX7), a subunit of canonical polycomb repressive complex 1 (PRC1), has been identified as a potent inducer of self-renewal in stem cells [11,12,13]. CBX7, in particular, displays strong affinity for the trimethylation of histone H3 on lysine 27 (H3K27me3) [14] and then downregulates p16 [15], a common cell cycle inhibitor. CBX7 also participates a positive auto-regulatory loop of Nanog expression, which favors multipotency maintenance [16]. Noteworthily, CBX7’ s known target p16 has been demonstrated to arrest angiogenesis in tumor progression via its impact on pro-angiogenic factors [17, 18]. From this point of view, CBX7 may also benefit stem cells in angiogenesis through its negative regulation of p16. Collectively, we assumed CBX7 as a promising molecule to rejuvenate late passage DPSCs by maintaining stemness and pro-angiogenic ability. A latest study unveiled that hypoxia-inducible factor-1α (HIF-1α) could directly upregulate CBX7 expression in neural progenitor cells (NPCs) and then intrinsically trigger NPC self-renewal [19]. This finding hinted that hypoxia may optimize regenerative potential of late passage DPSCs via sustainedly inducing CBX7 upregulation since early passage. Copper ion (Cu2+), a type of trace elements in humans [20], is well known to mimic hypoxia microenvironment by activating and stabilizing HIF-1α [21, 22]. So, we took Cu2+ for example to overexpress CBX7 in DPSCs via the initiation of HIF-1α-CBX7 cascade.
Here, we added Cu2+ to cell expansion medium of DPSCs since passage 3 (P3), and then investigated their proliferation, multipotency, odonto-differentiation and pro-angiogenic ability at passage 9 (P9) (Fig. 1). Through phenotypic analyses, we proposed a possible rejuvenation strategy to alleviate cell expansion-induced decline in stemness and angiogenesis, and this strategy may provide a powerful resource of self-renewing stem cells demanded for cell-based regeneration.

Schematic illustration for the effects of Cu2+-induced CBX7 overexpression on proliferation, multipotency, odonto-differentiation and pro-angiogenic ability of late passage DPSCs
2 Materials and methods
2.1 Cell culture
Human DPSCs (hDPSCs) were obtained from healthy donors aged between 18 and 25 years in Affiliated Stomatological Hospital of Nanjing Medical University. All experiments were approved by the Ethics Committee of Nanjing Medical University (No. 2021–177). Cells were cultured with complete medium (α-MEM with 10% fetal bovine serum, 100 μg/ml glutamine, 100 μg/ml penicillin and 100 μg/ml streptomycin) under an environment of 5% CO2 at 37 °C. hDPSCs at P3 and P9 were identified by flow cytometry using human mesenchymal stem cell (MSC) Analysis Kit (BD Biosciences, Bedford, MA, USA) and the results were shown in Fig. S1.
2.2 Cell viability assay
Cell viability was detected using cell-counting kit-8 (CCK-8; Dojido, Kumamoto, Japan). hDPSCs-P3 were seeded in 96-well plate at 2 × 105/ml and then cultured by medium supplemented with 0, 5, 25, 50, 100 μM Cu2+ diluted from a stock solution containing 50 mM copper sulfate (CuSO4 × 5H2O; Sigma-Aldrich, St. Louis, MO, USA) for 1, 3, 7 and 14 days. Cells were incubated with CCK-8 reagent at 37 °C for 1 h. We then measured the optical density values (OD) at a wavelength of 450 nm by a microplate reader (Bio-Rad, Hercules, CA, USA).
2.3 Chromatin immunoprecipitation (ChIP) assay
Directed by the manufacturer, we performed ChIP assays with EZ-ChIP (Millipore, Burlington, MA, USA). Generally, hDPSCs were fixed with 1% formaldehyde for 10 min at room temperature to crosslink proteins to DNA. The cells were then lysed and sonicated to shear crosslinked DNA to 200–1000 base pairs in length. DNA–protein complexes were immunoprecipitated with anti-HIF-1α (Cell Signaling Technology, Danvers, MA, USA) in combination with protein A magnetic beads at an overnight incubation. Purified DNA was then analyzed by qPCR using the CBX7 promoter primers based on a reported study (Forward: 5’-GCGTCTGGGCACCGACCACC-3’; Reverse: 5’-CCTAATCCGGCCTTCTCCGC-3’) [19].
2.4 Lentiviral transfection
In this study, recombinant lentivirus to overexpress CBX7 genes and relevant negative control lentivirus were purchased from GenePharma (GenePharma, Shanghai, China), while recombinant lentivirus to knockdown CBX7 genes (shCBX7: 5’-CGGAAGGGTAAAGTCGAGTAT-3’) and corresponding negative control lentivirus (shCtrl: 5’- TTCTCCGAACGTGTCACGTAA-3’) were purchased from HanBio Therapeutics (Shanghai, China). hDPSCs-P3 were plated in culture dishes at a density of 1.5 × 106 cells and cultured overnight. After reaching 60–70% confluence, the cells were transfected with the lentivirus (overexpression: MOI = 125, knockdown: MOI = 50). The medium was replaced with fresh complete medium after 24 h. CBX7 overexpressed/knockdown cells were harvested at 48 h for estimating transfection efficiency by western blot and subcultured for the following experiments.
2.5 Western blotting
hDPSCs from different groups were lysed by RIPA buffer (Beyotime, Shanghai, China) mixed with protease and phosphatase inhibitor cocktail (APExBIO, Houston, TX, USA). After being separated using 10% SDS–polyacrylamide gel, the total proteins (10 μg) were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore), which were then blocked in 5% nonfat milk for 1 h at room temperature, followed by incubating with anti-HIF-1α (1:1000, Proteintech, Rosemont, IL, USA), anti-CBX7 (1:1000, Proteintech) and anti-GAPDH (1:1000, Proteintech) overnight at 4 °C and then with horseradish peroxidase (HRP)-conjugated affinipure goat anti-mouse or rabbit IgG (1:10,000, Proteintech) for 1 h at room temperature. Finally, the protein bands were visualized by enhanced chemiluminescence kit (Vazyme, Shanghai, China). The intensity of bands was calculated by Image J software (National Institutes of Health, Bethesda, MD, USA).
2.6 Quantitive polymerase chain reaction (qPCR)
An RNA extraction kit (Bioteke, Wuxi, China) was used to extract total cellular RNA, which was then synthesized into cDNA by the HiScript III RT superMix for qPCR (Vazyme). The expressions of genes involved in proliferation, multipotency, angiogenesis and odontoblastic differentiation were detected using the ABI QuantStudio™ Real-Time PCR System (Applied Biosystems Bedford, MA, USA). The primer sequences used for this study are shown in Table 1. β-Actin was used as an internal control.
2.7 Cell immunofluorescence
hDPSCs were firstly fixed in 4% paraformaldehyde for 30 min before permeabilizing with 0.2% Triton X-100 (Sigma-Aldrich) for 5 min and blocking with goat serum (Boster Biological Technology, Wuhan, China) for 1 h. Then we incubated the cells with anti-Ki67 (1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-Nanog (1:100, Abcam, Cambridge, UK) antibodies at 4 °C overnight, followed with secondary antibodies (1:200, ABclonal Technology, Woburn, MA, USA) tagged with FITC or Cy3 for 40 min at room temperature. The nuclei of the cells were dyed with DAPI (Beyotime) and immunofluorescent images were taken with Leica DMI3000B (Leica, Wetzlar, Germany).
2.8 Mineral induction and ALP staining
hDPSCs were seeded into 12-well plates at a density of 5 × 104 cells per well. Upon reaching 60% confluency, cells were cultivated in mineral induction medium supplemented with 0.1 nM dexamethasone, 50 μg/ml ascorbate and 10 mM β-glycerophosphate (all from Sigma) for 7 days. The medium was changed every two days. After paraformaldehyde fixation, nitro blue tetrazolium (NBT) and 5-bromo-4-chroro-3-indolyl phosphate (BCIP) (Beyotime) were added in certain ratio to the cells for ALP staining in accordance with the manufacturer’ s instructions. Images of all the stained areas were acquired under a scanner.
2.9 Capillary-like tube formation
In order to examine the formation of capillary-like tubes, human umbilical vein endothelial cells (HUVECs) were utilized on Matrigel matrix (Corning, Corning, NY, USA) as previously described [23]. It was necessary to collect conditional media of each hDPSC groups after culturing for 24 h in α-MEM without serum prior to the experiment. Then the 96-well plates were coated with Matrigel and kept at 37 °C in a humidified environment for a minimum of 30 min. A total of resuspended 5 × 104 HUVECs per well were seeded on the surface of the Matrigel. Images of capillary tube formation were captured with an inverted microscope (Leica) and the meshes of the tubes were quantified using Image J software.
2.10 HUVECs migration
In the HUVECs migration assay, the lower chambers of 24-well plates with 8 μm transwell upper chambers (Millipore) was used for seeding hDPSCs. As soon as the cells achieved 90% confluence, HUVECs were seeded on the upper chamber with 200 μl serum-free endothelial cell medium (Sciencell Research Laboratories, Carlsbad, CA, USA) at a density of 5 × 103 cells per well. After 24 and 48 h of co-culture, the migrating cells on the lower surface of the transwell membrane were fixed with 4% paraformaldehyde and stained with Crystal Violet Staining Solution (Beyotime). Microscope images were taken to count the cells by a light microscope (Leica).
2.11 Cell viability in three-dimensional (3D) culture system
Hydrogels were prepared using plasminogen-depleted human fibrinogen (Sigma-Aldrich) solution diluted in serum-free α-MEM at a concentration of 10 mg/ml and filtered for sterility. hDPSCs-P9 were labeled by GFP using lentivirus prior to 3D culture. GFP-hDPSCs-P9 were then resuspended in prepared fibrinogen solution, totaling 3 × 104 cells per well of 24-well plate (500 μl total volume per well). 3 μl of thrombin solution (25 U/ml; Sigma-Aldrich) was added to 500 μl of fibrinogen-cell solution to catalyze gelation. Before fibrin polymerization, the plate was gently tapped on all four edges to guarantee the gel evenly distributed at the bottom of each well. After incubation at 37 °C for 30 min, gels were fed with 500 μl complete medium that was changed every three days. Fluorescent images were captured using a fluorescence inverted microscope at 1, 3 and 5 days.
2.12 Animal experiment
This study was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (No. 2005034–1). Eighteen male six-week-old BALB/c nude mice were used for this study. All mice were anaesthetized by intravenous injection of pentobarbital sodium (1.5 mg/kg) before surgery procedure. Two different murine models were fabricated in this study. As transport vehicle for hDPSCs in Model 1, plasminogen-depleted human fibrinogen solution was diluted by serum-free media at a concentration of 5 mg/ml and filtered for sterility. In Model 2, porous calcium phosphate cement (CPC; Rebone, Shanghai, China) with size Φ 4 mm × 2 mm was applied as cell scaffold for hDPSCs. Both P3 and P9 hDPSCs were used in two models. hDPSCs-P9+Cu2+ were generated from normal hDPSCs-P3 which were then cultured in complete medium with 25 μM Cu2+ until P9. The mesenchymal stemness markers of hDPSCs-P9+Cu2+ were detected by flow cytometric analysis and the results were shown in Fig. S1. Six mice were subject to Model 1 with injectants from three groups: hDPSCs-P9, hDPSCs-P9+Cu2+ and hDPSCs-P3. All hDPSCs were trypsinized and resuspended by fibrinogen solution with 5 × 105 hDPSCs per 100 μl. The fibrinogen-hDPSCs solution was catalyzed by thrombin solution (50 U/ml). Before completely gelation, hDPSCs-gel was quickly injected subcutaneously on the dorsal flank of the mouse. The three different groups were implanted once per mouse. After 14 days, the samples were collected and fixed in 4% paraformaldehyde. As for Model 2, hDPSCs-P9, hDPSCs-P9+Cu2+ and hDPSCs-P3 were all firstly cultured in mineral induction medium for 5 days. Then hDPSCs from each group were collected respectively and resuspended in serum-free medium with a concentration of 5 × 106 cells/ml. In subsequent surgical procedure, 200 μl cell suspention was added to each CPC implant. Twelve mice were subject to Model 2. Four subcutaneous pockets were created on the back of each mouse with a distance of more than 3 cm between each pocket, which randomly received the following implants of four groups: (1) CPC alone; (2) hDPSCs-P9/CPC complexes; (3) hDPSCs-P9+Cu2+/CPC complexes; (4) hDPSCs-P3/CPC complexes. Each six mice were sacrificed by euthanasia at 1 and 3 months after implantation, respectively. All implants were harvested and fixed in 4% paraformaldehyde, then decalcified in 10% EDTA.
2.13 Histological and immunohistochemical staining
All specimens were embedded in paraffin and sectioned at a thickness of 5 μm. Hematoxylin and eosin (HE) staining was processed as described previously [24]. Immunohistochemistry was performed using avidin–biotin–peroxidase complex technique with antibodies for CD31 (1:200, Santa Cruz Biotechnology), Ki67 (1:100, Proteintech), Sox-2 (1:200, Proteintech), DMP-1(1:200, Santa Cruz Biotechnology). In a nutshell, after dehydration, sections were treated with 3% H2O2 to remove endogenous hydrogen peroxide, blocked with goat serum and then incubated overnight with the primary antibodies listed above. Afterwards, sections were washed in PBS three times and incubated using MaxVision™ HRP-Polymer anti-Mouse/Rabbit IHC Kit (MXB, Fujian, China) and counterstained with DAB Plus Kit (MXB). Images were photographed with Leica DM4000 (Leica).
2.14 Statistical analysis
The significance of the experimental data was evaluated using two independent sample t-tests and one-way ANOVAs. All statistical analyses were conducted by the use of the GraphPad Prism 8.0 software. Results are presented as mean ± standard deviation (SD). *: p < 0.05, **: p < 0.01, ***: p < 0.001, ns: no significance.
3 Results
3.1 Cu2+ upregulated CBX7 expression through HIF-1α-CBX7 pathway in hDPSCs
Firstly, the toxicity of different Cu2+ concentrations to hDPSCs-P3 was measured by CCK-8 assay. On the first day, there were no significant differences in hDPSCs proliferation rate among different Cu2+ concentrations except for 100 μM. From day 3, hDPSCs cultured in 25 μM Cu2+ exhibited a remarkable increase in proliferative ability, compared with those in absence of Cu2+ or other concentrations of Cu2+. According to these results (Fig. 2A), 25 μM was chosen as the optimum Cu2+ concentration for the following experiments. To determine whether the expression of HIF-1α and CBX7 were increased under long-term stimulation of Cu2+ since P3, we detected their gene and protein levels in hDPSCs-P9. The data showed that both HIF-1α and CBX7 were highly expressed in hDPSCs-P9+Cu2+ group (Fig. 2C–G). To verify Cu2+-induced CBX7 upregulation through HIF-1α activation, we confirmed the recruitment of HIF-1α to the CBX7 gene promoter using ChIP assay in hDPSCs-P3 subjected to the stimulation of Cu2+ for 7 days. We did not observe this binding in hDPSCs-P3 cultured under normal conditions or in hDPSCs-P3 treated with both the pharmacological inhibitor 2-ME2 and Cu2+ (Fig. 2B).

Cu2+ upregulated CBX7 expression through HIF-1α-CBX7 pathway in hDPSCs. A CCK-8 assay detected the proliferation of hDPSCs-P3 cultured in 0, 5, 25, 50, 100 μM Cu2+. B ChIP assay verified the binding of HIF-1α to CBX7 promoter after Cu2+ stimulation for 7 days. HIF-1α expression was abolished by the pharmacological inhibitor 2-ME2. C–E Protein expression levels of HIF-1α and CBX7 in hDPSCs-P9 and hDPSCs-P9+Cu2+. F, G mRNA expression levels of HIF-1α and CBX7 in hDPSCs-P9 and hDPSCs-P9+Cu2+. Data expressed as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ns: no significance
3.2 Long-term stimulation of Cu2+ enhanced the proliferation and multipotency of late passage hDPSCs
Firstly, we evaluated the morphological changes of hDPSCs-P9 under long-term stimulation of Cu2+. As shown in Fig. 3A, normal hDPSCs-P9 displayed the enlarged cell bodies and elongated appearance compared with hDPSCs-P9+Cu2+ group. To investigate whether long-term stimulation of Cu2+ increased the proliferation of hDPSCs-P9, we evaluated Ki67 by immunocytochemistry and found that Cu2+ significantly increased Ki67 immunostaining in hDPSCs-P9 (Fig. 3B, D). As a known target of CBX7, the expression of p16 was decreased in hDPSCs-P9+Cu2+ group following Cu2+-induced CBX7 overexpression (Fig. 3F). Although the proliferative ability of hDPSCs-P9+Cu2+ was lower than normal hDPSCs-P3, it notably exceeded that of normal hDPSCs-P9. We then analyzed the expression of multipotency markers in hDPSCs. Compared with normal hDPSCs-P9, Nanog, Oct-4 and Sox-2 were significantly upregulated in hDPSCs-P9+Cu2+ group as shown by qPCR (Fig. 3G–I). The immunofluorescent staining of Nanog (Fig. 3C, E) exhibited the same trend as the qPCR results. Notwithstanding the weakness in comparison of hDPSCs-P3, hDPSCs-P9+Cu2+ displayed enhanced self-renewal than normal hDPSCs-P9.

Long-term stimulation of Cu2+ enhanced the proliferation and multipotency of late passage hDPSCs. A Inverted microscopy images of hDPSCs-P9 and hDPSCs-P9+Cu2+ (Scale bar: 200 μm). B, D Immunofluorescence detection of Ki67 expression (Scale bar: 200 μm). C, E Immunofluorescence detection of Nanog expression (Scale bar: 200 μm). F–I mRNA expression levels of p16, Nanog, Oct-4 and Sox-2 in hDPSCs-P9, hDPSCs-P9+Cu2+ and hDPSCs-P3. Data expressed as mean ± SD. *: p < 0.05, ***: p < 0.001, ns: no significance
3.3 Long-term stimulation of Cu2+ promoted the odontogenic differentiation and angiogenesis of late passage hDPSCs in vitro
To determine whether increased self-renewal potency of hDPSCs-P9+Cu2+ could facilitate the odontogenic differentiation, we performed ALP staining after mineral induction for 7 days. Staining was more intense in hDPSCs-P9+Cu2+ than normal hDPSCs-P9 (Fig. 4A, B). Consistently, the odontoblast-lineage marker DSPP, which plays an important role in dentin mineralization, was significantly upregulated in hDPSCs-P9+Cu2+ compared with normal hDPSCs-P9 (Fig. 4C). As is known, Cu2+ can induce angiogenesis via increasing the expression of pro-angiogenic factors, like VEGF [25]. As a downstream molecule of Cu2+, CBX7 may also facilitate the secretion of angiogenesis-related factors via its target p16. Hence, we detected the gene level of VEGF and found it markedly increased in hDPSCs-P9+Cu2+ compared to normal hDPSCs-P9 (Fig. 4F). Considering VEGF secreted by hDPSCs may enhance pro-angiogenic ability, we then performed angiogenesis assay. HUVECs were seeded into 96-well plates coated with Matrigel, and cultured with conditional media collected from different groups respectively. After 6 h culture, significantly increased number of tube-like structures was observed in hDPSCs-P9+Cu2+ group compared with hDPSCs-P9 group (Fig. 4D, G). When hDPSCs-P9+Cu2+ were co-cultured with HUVECs for 24 and 48 h, more HUVECs migrated across the transwell membrane than those co-cultured with normal hDPSCs-P9 at both time points (Fig. 4E, H, I). Collectively, these findings revealed enhanced odontoblastic differentiation and pro-angiogenesis of hDPSCs-P9 under long-term stimulation of Cu2+, although these abilities were still inferior to those of hDPSCs-P3.

Long-term stimulation of Cu2+ promoted the odontogenic differentiation and angiogenesis of late passage hDPSCs in vitro. A, B ALP staining of hDPSCs from different groups and the positive stained area analysis. C mRNA expression levels of DSPP in hDPSCs-P9, hDPSCs-P9+Cu2+ and hDPSCs-P3. D, G Capillary-like tube formation assay of HUVECs cultured with conditional media from different groups and the tube-like numbers of HUVEC-sprouting analysis (Scale bar: 200 μm). E, H, I Migration assay of hDPSCs from different groups at 24 and 48 h (Scale bar: 200 μm). F mRNA expression levels of VEGF in hDPSCs-P9, hDPSCs-P9+Cu2+ and hDPSCs-P3. Data expressed as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ns: no significance
3.4 Cu2+ facilitated local vascularization and self-renewing capacity of late passage hDPSCs subcutaneously injected in hydrogel
As a common cell delivery vehicle, hydrogel serves as a provisional extracellular matrix that closely recapitulates cellular microenvironment in vivo [26]. We hereby applied hydrogel as the transport device of hDPSCs in a subcutaneous injection model of nude mice. Firstly, we determined the gelation rate of hDPSCs-hydrogel and examined the viability of hDPSCs cultured in hydrogel. As shown in Fig. 5A, hydrogel semi-gelated at 90 s, completely gelated at 120 s, and hDPSCs could grow and proliferate well in hydrogel. These results demonstrated hydrogel suitable for the subsequent experiments. After 2 weeks, the histological results showed that hydrogel was completely absorbed in all groups (Fig. 5E). Immunohistochemical examination of CD31, Ki67 and Sox-2 were then performed to investigate the effects of long-term stimulation of Cu2+ on the local vascularization and self-renewing capacity of late passage hDPSCs in vivo. In accordance with in vitro results, the expression of all three markers was remarkably upregulated in hDPSCs-P9+Cu2+ group but barely detectable in hDPSCs-P9 group (Fig. 5B–E). Although slightly lower than those of hDPSCs-P3, the pro-angiogenic ability and self-renewing capacity of hDPSCs-P9+Cu2+ were significantly enhanced in local transplanted microenvironment within the first two weeks.

Cu2+ facilitated local vascularization and self-renewing capacity of late passage hDPSCs subcutaneously injected in hydrogel. A Schematic illustration of hDPSCs-hydrogel subcutaneous injection (Scale bar: 200 μm). B–D Percentage of CD31, Ki67 and Sox-2 positive area in different groups. E HE staining and CD31, Ki67 and Sox-2 immunohistochemical staining of subcutaneous tissue from different groups after hDPSCs-hydrogel subcutaneous injection for 2 weeks (Scale bar: HE, 200 μm; Immunohistochemical staining, 100 μm for CD31, and 50 μm for Ki67 and Sox-2). Data expressed as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001
3.5 Cu2+ boosted the odontoblastic differentiation of late passage hDPSCs subcutaneously transplanted with CPC
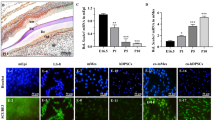
To determine the effects of Cu2+ on the odontoblastic differentiation of late passage hDPSCs in vivo, we mixed hDPSCs from each group with porous CPC scaffolds, and then transplanted CPC alone/hDPSCs-CPC complexes subcutaneously into nude mice for 1 and 3 months (Fig. 6A). After 1 month, a small amount of mineralized tissue was found in hDPSCs-P9+Cu2+/CPC and hDPSCs-P3/CPC complexes, while little or no mineralized tissue was formed in hDPSCs-P9/CPC complexes or CPC alone (Fig. 6B, E). When the implantation time was extended to 3 months, there was more mineralized tissue in all three hDPSCs/CPC groups, and significantly more mineralized tissue was observed in hDPSCs-P9+Cu2+ and hDPSCs-P3 groups (Fig. 6C, E). At both month1 and 3, slightly more mineralized tissue was formed in hDPSCs-P3 group than in hDPSCs-P9+Cu2+ group. The DMP-1 immunohistochemical analysis further demonstrated the odontoblastic differentiation of hDPSCs in different groups. As shown in Fig. 6D, E a DMP-1 positive layer lined the inner surface of newly formed mineralized tissue in hDPSCs-P9+Cu2+ group, and the positive staining layer was more apparent in hDPSCs-P3 group but almost undetectable in hDPSCs-P9 group. Despite lower-than-normal hDPSCs-P3, hDPSCs-P9+Cu2+ exhibited markedly enhanced odontoblastic differentiation capacity compared with normal hDPSCs-P9.

Cu2+ boosted the odontoblastic differentiation of late passage hDPSCs subcutaneously transplanted with CPC. A Schematic illustration of the procedure for transplantation of CPC alone/hDPSCs-CPC complexes into nude mice. B–D Percentage of mineralized and DMP-1 positive area in different groups. E HE staining and DMP-1 immunohistochemical staining of CPC alone/hDPSCs-CPC complexes after 1 month and 3 months subcutaneous transplantation (Scale bar: HE, 200 μm; Immunohistochemical staining for DMP-1, 100 μm). Data expressed as mean ± SD. *: p < 0.05, ***: p < 0.001
3.6 CBX7 was the key factor to maintain stemness of late passage hDPSCs
To determine whether the effects of Cu2+ on late passage hDPSCs were mainly dependent on the overexpression of CBX7, we used lentiviral CBX7 short hairpin RNA (LV-CBX7-sh) to abolish the normal expression of CBX7 in hDPSCs, meanwhile, established CBX7 overexpressed hDPSCs by CBX7-induced lentiviral (LV-CBX7) infection (Fig. 7A, B). As shown in Fig. 7C–J, CBX7 overexpression maintained the proliferation and multipotency of hDPSCs-P9 almost as strong as those of hDPSCs-P3. Conversely, the abrogation of CBX7 expression lowered the normal level of hDPSCs-P9 on proliferation and multipotency. As crucial marker genes involved in odontoblastic differentiation, BSP, DMP-1 and DSPP were significantly increased in P9-CBX7+/+ but markedly decreased in P9-CBX7−/− in contrast to normal P9 (Fig. 7M–O). ALP staining was consistent with the qPCR results (Fig. 7K, L). These results showed that the odonto-differentiation capacity was almost the same between CBX7 overexpressed hDPSCs-P9 and normal hDPSCs-P3, but it would be impaired in normal hDPSCs-P9 subsequent to the downregulation of CBX7.

CBX7 was the key factor to maintain stemness of late passage hDPSCs. A Fluorescence microscopy images of LV-CBX7-sh and LV-control-sh transfected hDPSCs and protein expression levels of CBX7 displaying knockdown efficiency (Scale bar: 100 μm). B Fluorescence microscopy image of LV-CBX7 transfected hDPSCs and protein expression levels of CBX7 displaying overexpression efficiency (Scale bar: 100 μm). C, E Immunofluorescence detection of Ki67 expression in different groups (Scale bar: 200 μm). D, F Immunofluorescence detection of Nanog expression in different groups (Scale bar: 200 μm). G-J mRNA expression levels of p16, Nanog, Oct-4 and Sox-2 in P9, P9-CBX7−/−, P9-CBX7+/+ and P3. K, L ALP staining of different groups and the positive stained area analysis. M–O mRNA expression levels of BSP, DMP-1 and DSPP in different groups. Data expressed as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ns: no significance
3.7 CBX7 prompted pro-angiogenic ability of late passage hDPSCs
To verify an independent role of CBX7 in prompting pro-angiogenic ability of late passage hDPSCs, we firstly detected the expression of VEGF in gene level, and found it upregulated in P9-CBX7+/+ but decreased in P9-CBX7−/− compared to normal P9 (Fig. 8C). Next, we performed HUVECs capillary-like tube formation assay. HUVECs cultured with conditional media from P9-CBX7+/+ group formed more capillary-like tubes on Matrigel after 6 h, while tube structures were barely observed in P9-CBX7−/− group in contrast to normal P9 group (Fig. 8A, D). CBX7 could also induce HUVECs to migrate across the transwell membrane. After 24 h co-culture, the number of HUVECs in field of P9-CBX7+/+ group was higher than that in normal P9 group, while markedly less HUVECs migrated in P9-CBX7−/− group in contrast to normal P9 group (Fig. 8B, E). The same trend could be observed after 48 h co-culture (Fig. 8B, F). Although lower than that of normal P3, the pro-angiogenic ability of P9-CBX7+/+ were stronger than that of normal P9, and it would be weakened due to CBX7 knockdown.

CBX7 prompted pro-angiogenic ability of late passage hDPSCs. A, D Capillary-like tube formation assay of HUVECs cultured with conditional media from different groups and the tube-like numbers of HUVEC-sprouting analysis at 6 h (Scale bar: 200 μm). B, E, F Migration assay of hDPSCs from different groups at 24 and 48 h (Scale bar: 200 μm). C mRNA expression levels of VEGF in P9, P9-CBX7−/−, P9-CBX7+/+ and P3. Data expressed as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001
4 Discussion
Advances in stem cell biology have aided stem cell-based tissue regeneration. DPSCs are well-documented as a viable source for tissue regeneration due to their multipotential capabilities [27, 28]. Although DPSCs are relatively accessible, the population of self-renewable stem cells could hardly satisfy the clinical use. Reportedly, the proportion of DPSCs with high proliferation and ability to regenerate ectopic dentin in vivo is very low [4, 29] among DPSCs isolated from pulp tissue via the explant method [30, 31] or the enzymatic digestion method [32]. Moreover, extensive in vitro cell expansion eventually leads to proliferative decline and impaired oriented/spontaneous differentiation ability [33]. Hence, stemness maintenance has emerged as a challenging topic in stem cell therapy, owing to insufficient supply and decreased stemness after long-term in vitro culture. To date, stemness maintenance can be realized by adding growing factors/drugs to culture medium, such as fibroblast growth factor (bFGF) [34,35,36], epidermal growth factor (EGF) [37], melatonin [38], and l-ascorbate 2-phosphate (A2-P) [39], or applying nanomaterials as cell scaffolds, like graphene-coated substrates [40], and ZnO nanorod arrays [41]. However, high cost, easy degradability, and complicated fabrication process greatly limit the application of these strategies.
Stemness is the capability of undifferentiated cells to undergo an indefinite number of replications (self-renewal) and give rise to specialized cells (differentiation) [42]. CBX7 has been widely reported as a key factor in sustaining self-renewal potential of various stem cells. As a "reader" of H3K27me3 marker [43], CBX7 competitively interacts with H3K27me3 instead of other CBX proteins to safeguard self-renewal of stem cells, including repressing lineage-specific markers and downregulating p16 [13, 44]. Besides, CBX7 cooperates with Nanog at the distal upstream region of Nanog to form a positive auto-regulatory loop of Nanog expression, which benefits multipotency maintenance [16]. So, we detected proliferation/multipotency-related markers (including p16 and Nanog) and found that CBX7 overexpression maintained the proliferative and multipotential capabilities of hDPSCs-P9 almost as strong as those of hDPSCs-P3. Considering another aspect of stemness, we also analyzed changes in odonto-differentiation of hDPSCs in vitro, and discovered that both gene level of odontoblast-lineage markers and calcium precipitation were nearly the same between P9-CBX7+/+ and normal P3. These results collectively identify CBX7 as a crucial factor in stemness maintenance. Restricted by impaired stemness accompanying with cell expansion, hMSCs cultures in vitro are recommended in cell therapy up to passage 4 [45]. Nevertheless, our findings of CBX7 may break this limitation and provide a powerful resource of stem cells for cell-based regeneration. A latest study uncovered that HIF-1α-CBX7 pathway play a considerable role in stem cell self-renewal [19]. As is known, Cu2+ activates and stabilizes HIF-1α [21, 22]. Thus, we applied Cu2+ to achieve CBX7 overexpression in hDPSCs via triggering HIF-1α-CBX7 cascade. In this study, we firstly demonstrated Cu2+-activated HIF-1α directly bound to CBX7 promoter to upregulate CBX7 expression in hDPSCs. Next, we added Cu2+ to cell expansion medium since P3, and found long-term stimulation of Cu2+ both enhance the proliferation and multipotency of hDPSCs-P9 and boost the odontogenic differentiation of hDPSCs-P9 under mineral induction. From this point of view, adding Cu2+ to cell expansion system may serve as a new strategy to maintain stemness for its low expense, simple operation and well performance in retarding cell expansion-induced decrease of stemness.
Recent studies have indicated that the therapeutic benefits of MSCs is attributable to not only their stemness but also factors they secrete [2]. DPSCs have been reported to induce angiogenesis partially by their paracrine property [5]. Like other MSCs, DPSCs are conditioned by the interaction with hypoxia microenvironment and secrete a mass of VEGF, the master regulator of vascular growth [46, 47]. To data, anti-VEGF/anti-angiogenesis function of p16 (a known target of CBX7) has been proved to suppress tumor growth [17], but no studies have documented a direct and positive effect of CBX7 on pro-angiogenic ability of stem cells. Hence, we detected the gene level of VEGF in hDPSCs-P9, and found it upregulated with CBX7 overexpression but significantly downregulated with CBX7 knockdown. In angiogenesis assay, the number of capillary-like structures and migrating co-cultured HUVECs was increased in P9-CBX7+/+ group but markedly decreased in P9-CBX7−/−group. These findings revealed that CBX7 may directly enhance pro-angiogenic ability of stem cells through upregulating VEGF expression. This mechanism involves in the classical function of Cu2+ on the activation of HIF-1α-VEGF pathway, but the role of CBX7 in this pathway has not yet been reported. As a novel downstream target of HIF-1α, does CBX7 serve as a pivotal link between HIF-1α and VEGF? In this perspective, our data provide an insightful view into the mechanisms underlying Cu2+-induced promotion of angiogenesis.
Last but not least, we detected the safety of Cu2+ addition to long-term cell expansion medium. On one hand, flow cytometric analysis of hDPSCs-P9+Cu2+ for mesenchymal stemness markers indicated stem cell phenotype of hDPSCs insusceptible to long-term cultivation under Cu2+. On the other hand, histological and immunohistochemical staining of subcutaneously injected hDPSCs-P9+Cu2+ displayed good survival and low tumorigenicity in vivo, considering Ki67 expression of hDPSCs-P9+Cu2+ still slightly lower than that of normal hDPSCs-P3. So far, the introduction of Cu2+ to stem cell microenvironment has been well studied in the field of cell-based regeneration [48], but little is known about the influence of Cu2+-containing long-term culture on stem cells. Therefore, we investigated changes of stem cell properties in response to long-term stimulation of Cu2+, which has no adverse effect on stem cells.
Taken together, our data indicate CBX7 as an effective factor to maintain stemness and pro-angiogenic ability of stem cells insusceptible to cell expansion. We used Cu2+ to achieve CBX7 overexpression through initiating HIF-1α-CBX7 cascade, and found ameliorated regenerative potential of late passage stem cells both in vitro and in vivo. Given the safety of Cu2+ addition to long-term cell expansion medium, we propose a new strategy with low expense and simple operation to manufacture large population of stem cells for fulfilling the demand for cell-mediated tissue regeneration.
References
Kolf CM, Cho E, Tuan RS. Mesenchymal stromal cells. Biology of adult mesenchymal stem cells: regulation of niche, self-renewal and differentiation. Arthritis Res Ther. 2007;9:204.
Madrigal M, Rao KS, Riordan NH. A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J Transl Med. 2014;12:260.
Wagner W, Ho AD, Zenke M. Different facets of aging in human mesenchymal stem cells. Tissue Eng Part B Rev. 2010;16:445–53.
Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, et al. Stem cell properties of human dental pulp stem cells. J Dent Res. 2002;81:531–5.
Merckx G, Hosseinkhani B, Kuypers S, Deville S, Irobi J, Nelissen I, et al. Angiogenic effects of human dental pulp and bone marrow-derived mesenchymal stromal cells and their extracellular vesicles. Cells. 2020;9:312.
Alge DL, Zhou D, Adams LL, Wyss BK, Shadday MD, Woods EJ, et al. Donor-matched comparison of dental pulp stem cells and bone marrow-derived mesenchymal stem cells in a rat model. J Tissue Eng Regen Med. 2010;4:73–81.
Zhang W, Walboomers XF, van Osch GJ, van den Dolder J, Jansen JA. Hard tissue formation in a porous HA/TCP ceramic scaffold loaded with stromal cells derived from dental pulp and bone marrow. Tissue Eng Part A. 2008;14:285–94.
Giuliani A, Manescu A, Langer M, Rustichelli F, Desiderio V, Paino F, et al. Three years after transplants in human mandibles, histological and in-line holotomography revealed that stem cells regenerated a compact rather than a spongy bone: biological and clinical implications. Stem Cells Transl Med. 2013;2:316–24.
d’Aquino R, De Rosa A, Lanza V, Tirino V, Laino L, Graziano A, et al. Human mandible bone defect repair by the grafting of dental pulp stem/progenitor cells and collagen sponge biocomplexes. Eur Cell Mater. 2009;18:75–83.
Xuan K, Li B, Guo H, Sun W, Kou X, He X, et al. Deciduous autologous tooth stem cells regenerate dental pulp after implantation into injured teeth. Sci Transl Med. 2018;10:eaaf3227.
Jung J, Buisman SC, Weersing E, Dethmers-Ausema A, Zwart E, Schepers H, et al. CBX7 induces self-renewal of human normal and malignant hematopoietic stem and progenitor cells by canonical and non-canonical interactions. Cell Rep. 2019;26:1906-18.e8.
Fan JR, Lee HT, Lee W, Lin CH, Hsu CY, Hsieh CH, et al. Potential role of CBX7 in regulating pluripotency of adult human pluripotent-like olfactory stem cells in stroke model. Cell Death Dis. 2018;9:502.
Ni SJ, Zhao LQ, Wang XF, Wu ZH, Hua RX, Wan CH, et al. CBX7 regulates stem cell-like properties of gastric cancer cells via p16 and AKT-NF-κB-miR-21 pathways. J Hematol Oncol. 2018;11:17.
Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol Cell Biol. 2006;26:2560–9.
Gil J, Bernard D, Martinez D, Beach D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat Cell Biol. 2004;6:67–72.
Hu S, Huo D, Yu Z, Chen Y, Liu J, Liu L, et al. ncHMR detector: a computational framework to systematically reveal non-classical functions of histone modification regulators. Genome Biol. 2020;21:48.
Lu Y, Zhang X, Zhang J. Inhibition of breast tumor cell growth by ectopic expression of p16/INK4A via combined effects of cell cycle arrest, senescence and apoptotic induction, and angiogenesis inhibition. J Cancer. 2012;3:333–44.
Baruah P, Lee M, Wilson PO, Odutoye T, Williamson P, Hyde N, et al. Impact of p16 status on pro- and anti-angiogenesis factors in head and neck cancers. Br J Cancer. 2015;113:653–9.
Chiu HY, Lee HT, Lee KH, Zhao Y, Hsu CY, Shyu WC. Mechanisms of ischaemic neural progenitor proliferation: a regulatory role of the HIF-1α-CBX7 pathway. Neuropathol Appl Neurobiol. 2020;46:391–405.
Rodríguez JP, Ríos S, González M. Modulation of the proliferation and differentiation of human mesenchymal stem cells by copper. J Cell Biochem. 2002;85:92–100.
Feng W, Ye F, Xue W, Zhou Z, Kang YJ. Copper regulation of hypoxia-inducible factor-1 activity. Mol Pharmacol. 2009;75:174–82.
Martin F, Linden T, Katschinski DM, Oehme F, Flamme I, Mukhopadhyay CK, et al. Copper-dependent activation of hypoxia-inducible factor (HIF)-1: implications for ceruloplasmin regulation. Blood. 2005;105:4613–9.
Jiang F, Zhang W, Zhou M, Zhou Z, Shen M, Chen N, et al. Human amniotic mesenchymal stromal cells promote bone regeneration via activating endogenous regeneration. Theranostics. 2020;10:6216–30.
Miao DS, He B, Karaplis AC, Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J Clin Investig. 2002;109:1173–82.
Bharathi Devi SR, Dhivya MA, Sulochana KN. Copper transporters and chaperones: their function on angiogenesis and cellular signalling. J Biosci. 2016;41:487–96.
Carrion B, Janson IA, Kong YP, Putnam AJ. A safe and efficient method to retrieve mesenchymal stem cells from three-dimensional fibrin gels. Tissue Eng Part C Methods. 2013;20:252–63.
Tatullo M, Marrelli M, Shakesheff KM, White LJ. Dental pulp stem cells: function, isolation and applications in regenerative medicine. J Tissue Eng Regen Med. 2015;9:1205–16.
Alraies A, Alaidaroos NY, Waddington RJ, Moseley R, Sloan AJ. Variation in human dental pulp stem cell ageing profiles reflect contrasting proliferative and regenerative capabilities. BMC Cell Biol. 2017;18:12.
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A. 2000;97:13625–30.
Spath L, Rotilio V, Alessandrini M, Gambara G, De Angelis L, Mancini M, et al. Explant-derived human dental pulp stem cells enhance differentiation and proliferation potentials. J Cell Mol Med. 2010;14:1635–44.
Hilkens P, Gervois P, Fanton Y, Vanormelingen J, Martens W, Struys T, et al. Effect of isolation methodology on stem cell properties and multilineage differentiation potential of human dental pulp stem cells. Cell Tissue Res. 2013;353:65–78.
Laino G, Graziano A, d’Aquino R, Pirozzi G, Lanza V, Valiante S, et al. An approachable human adult stem cell source for hard-tissue engineering. J Cell Physiol. 2006;206:693–701.
Izadpanah R, Kaushal D, Kriedt C, Tsien F, Patel B, Dufour J, et al. Long-term in vitro expansion alters the biology of adult mesenchymal stem cells. Cancer Res. 2008;68:4229–38.
Park SB, Yu KR, Jung JW, Lee SR, Roh KH, Seo MS, et al. bFGF enhances the IGFs-mediated pluripotent and differentiation potentials in multipotent stem cells. Growth Factors. 2009;27:425–37.
Wu J, Huang GT, He W, Wang P, Tong Z, Jia Q, et al. Basic fibroblast growth factor enhances stemness of human stem cells from the apical papilla. J Endod. 2012;38:614–22.
Tasso R, Gaetani M, Molino E, Cattaneo A, Monticone M, Bachi A, et al. The role of bFGF on the ability of MSC to activate endogenous regenerative mechanisms in an ectopic bone formation model. Biomaterials. 2012;33:2086–96.
Sabri A, Ziaee AA, Ostad SN, Alimoghadam K, Ghahremani MH. Crosstalk of EGF-directed MAPK signalling pathways and its potential role on EGF-induced cell proliferation and COX-2 expression in human mesenchymal stem cells. Cell Biochem Funct. 2011;29:64–70.
Shuai Y, Liao L, Su X, Yu Y, Shao B, Jing H, et al. Melatonin treatment improves mesenchymal stem cells therapy by preserving stemness during long-term in vitro expansion. Theranostics. 2016;6:1899–917.
Yu J, Tu YK, Tang YB, Cheng NC. Stemness and transdifferentiation of adipose-derived stem cells using L-ascorbic acid 2-phosphate-induced cell sheet formation. Biomaterials. 2014;35:3516–26.
Chen GY, Pang DW, Hwang SM, Tuan HY, Hu YC. A graphene-based platform for induced pluripotent stem cells culture and differentiation. Biomaterials. 2012;33:418–27.
Kong Y, Ma B, Liu F, Chen D, Zhang S, Duan J, et al. Cellular stemness maintenance of human adipose-derived stem cells on ZnO nanorod arrays. Small. 2019;15:e1904099.
La Noce M, Paino F, Spina A, Naddeo P, Montella R, Desiderio V, et al. Dental pulp stem cells: State of the art and suggestions for a true translation of research into therapy. J Dent. 2014;42:761–8.
O’Loghlen A, Muñoz-Cabello AM, Gaspar-Maia A, Wu HA, Banito A, Kunowska N, et al. MicroRNA regulation of Cbx7 mediates a switch of Polycomb orthologs during ESC differentiation. Cell Stem Cell. 2012;10:33–46.
Morey L, Pascual G, Cozzuto L, Roma G, Wutz A, Benitah Salvador A, et al. Nonoverlapping functions of the polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell. 2012;10:47–62.
Binato R, de Souza FT, Lazzarotto-Silva C, Du Rocher B, Mencalha A, Pizzatti L, et al. Stability of human mesenchymal stem cells during in vitro culture: considerations for cell therapy. Cell Prolif. 2013;46:10–22.
Bronckaers A, Hilkens P, Fanton Y, Struys T, Gervois P, Politis C, et al. Angiogenic properties of human dental pulp stem cells. PLoS One. 2013;8:e71104.
Mattei V, Martellucci S, Pulcini F, Santilli F, Sorice M, Delle MS. Regenerative potential of dpscs and revascularization: direct, paracrine or autocrine effect? Stem Cell Rev Rep. 2021;17:1635–46.
Wu C, Zhou Y, Xu M, Han P, Chen L, Chang J, et al. Copper-containing mesoporous bioactive glass scaffolds with multifunctional properties of angiogenesis capacity, osteostimulation and antibacterial activity. Biomaterials. 2013;34:422–33.
Acknowledgements
This study was jointly supported by the National Natural Science Foundation of China (81900960, 82270955), the Natural Science Foundation of Jiangsu Province (BK20200667, BK20221302), the Natural Science Foundation of Jiangsu Higher Education Institutions (20KJD320005) and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD, 2018-87). The authors indicated no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare no potential conflicts of interest.
Ethical statement
The study was approved by the Ethics Committee of Nanjing Medical University (No. 2021–177). The animal experiment was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (No. 2005034–1).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, Y., Li, B., Yu, D. et al. CBX7 Rejuvenates Late Passage Dental Pulp Stem Cells by Maintaining Stemness and Pro-angiogenic Ability. Tissue Eng Regen Med 20, 473–488 (2023). https://doi.org/10.1007/s13770-023-00521-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13770-023-00521-4