
Abstract
Background:
Angiogenesis and vasculogenesis are essential processes for successful tissue regeneration in tissue engineering and regenerative medicine. The adipose-derived stromal vascular fraction (SVF) is not only a source of adipose stem cells (ASC) but also a suitable source of microvascular endothelial cells because it is a rich capillary network. So, we propose a new hypothesis for isolating adipose-derived human microvascular endothelial cells (HMVEC-A) from the SVF and developed a dual isolation system that isolates two cell types from one tissue.
Method:
To isolate HMVEC-A, we analyzed the supernatant discarded when ASC is isolated from the adipose-derived SVF. Based on this analysis, we assumed that the SVF adherent to the bottom of the culture plate was divided into two fractions: the stromal fraction as the ASC-rich fraction, and the vascular fraction (VF) as the endothelial cells-rich fraction floating in the culture supernatant. VF isolation was optimized and the efficiency was compared, and the endothelial cells characteristics of HMVEC-A were confirmed by flow cytometric analysis, immunocytochemistry (ICC), a DiI-acetylated low-density lipoprotein (DiI-Ac-LDL) uptake, and in vitro tube formation assay.
Results:
Consistent with the hypothesis, we found a large population of HMVEC-A in the VF and isolated these HMVEC-A by our isolation method. Additionally, this method had higher yields and shorter doubling times than other endothelial cells isolation methods and showed typical morphological and phenotypic characteristics of endothelial cells.
Conclusion:
Cells obtained by the method according to our hypothesis can be applied as a useful source for studies such as tissue-to-tissue networks, angiogenesis and tissue regeneration, patient-specific cell therapy, and organoid chips.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Tissue engineering and regenerative medicine basically require a variety of cells. Among them, endothelial cells deliver an appropriate level of nutrients and oxygen through angiogenesis, and play a role in removing waste products, so proper blood vessel formation is an important system for successful tissue regeneration [1,2,3]. In addition, the need for endothelial cells is increasing for patient-specific cell therapy, organoid chips, disease modeling, drug screening, and clinical applications [4,5,6,7]. A number of cell isolation methods have been studied in various tissues to obtain functional cells that are well suited to patients immunologically and cells with desired biological properties, and adipose tissue among various tissues is an optimized source for obtaining various cells in this situation [8, 9]. Adipose tissue is easily obtained during liposuction or breast reconstruction and consists of various types of cells, including mature adipocytes, fibroblasts, adipocyte progenitors, and a range of inflammatory leukocytes. It is also a viable source of microvascular endothelial cells due to its abundant network of capillaries [8, 10, 11]. A stromal vascular fraction (SVF) with potential tissue regeneration activity is defined as a heterogeneous population of freshly isolated cells from adipose tissue [12]. The SVF is a heterogeneous mixture of 2 to 10% adipose stem cells (ASC), 7 to 30% endothelial cells (EC) (mature and progenitor cells), and up to 50% fibroblasts [13,14,15,16]. The vascular endothelial cells in the SVF are microvascular cells involved in angiogenesis, wound healing, and inflammation and are essential for the repair and reconstitution of damaged tissue [17,18,19]. To isolate ASC from this heterogeneous mixture of the SVF, ASC is harvested from the population of cells adhered to the bottom of the culture plate after a certain incubation time. During ASC isolation, the supernatant is discarded during the washing process to obtain the ASC attached to the bottom of the culture plate. We assumed that this discarded supernatant contains cells other than ASC and proposed a new hypothesis accordingly. First, we assumed that the SVF is divided into a stromal fraction (SF), an ASC-rich fraction initially adherent to the bottom of the culture plate, and a vascular fraction (VF), an EC-rich fraction floating in the supernatant (Fig. 1A). That is, the SF is the fraction in which ASC is isolated by adhesion to the bottom of the culture plate after primary incubation of the SVF, and the VF is the fraction in which a large number of endothelial cells are suspended in the supernatant after primary incubation of the SVF. Therefore, the purpose of this study was to develop a method for efficiently isolating large numbers of adipose-derived human microvascular endothelial cells (HMVEC-A) from discarded adipose tissue-derived SVF. We report a double harvesting protocol for obtaining two types of cells. This method is standardized, reliable, and easy to perform for the isolation of HMVEC-A while optimizing the isolation of ASC and HMVEC-A from fresh adipose tissue. Moreover, it is designed to shorten the isolation procedure based on the plastic adhesion method for cell isolation using ASC and pericytes [20,21,22,23]. Based on our hypothesis, we determined the optimal time for harvesting the VF and compared the isolated HMVEC-A with HMVEC-A isolated by the most commonly used methods, fluorescence-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS). In addition, we aimed to characterize HMVEC-A obtained by the optimal VF isolation method.

Hypothesis and schematic of the method for isolating HMVEC-A from adipose tissue. A We hypothesized that the stromal vascular fraction (SVF) is divided into the adipose-derived stem cell (ASC)-rich stromal fraction (SF) adherent to the bottom of the culture plate and the endothelial cell (EC)-rich vascular fraction (VF) floating in the supernatant. Isolation of cells from the supernatant, which excludes cells adhered to the bottom of the plate (ASC), is a VF-based isolation method that is performed later to isolate suspended cells (i.e., HMVEC-A). B Schematic showing the double harvesting system used to obtain HMVEC-A. The schematic shows the ASC adhesion step and the VF isolation step from the adipose-derived SVF
2 Materials and methods
2.1 Establishment of HUVEC and HMVEC cell lines as positive controls for HMVEC-A
Human umbilical vein endothelial cells (HUVEC, Lonza, Walkersville, MD, USA), and human microvascular endothelial cells (HMVEC, Lonza) were used as positive endothelial cells controls in the analysis of HMVEC-A. These cells were used according to the protocol approved by Lonza Inc.
2.2 Method for isolating microvascular endothelial cells from human adipose tissue
Informed consent was obtained from subjects in accordance with the regulations of the Institutional Review Board of Seoul St Mary's Hospital, Seoul, Korea.
Human adipose tissue was obtained from tissue discarded after liposuction or resection of fat tissue. The obtained samples were washed with phosphate-buffered saline (PBS; Wisent Inc., Quebec, Canada) and digested with 0.1% collagenase type I (Sigma-Aldrich, St. Louis, MO, USA) in a Celltibator (Medikan, Seoul, Korea) for 30 min at 37 °C. The digested samples were washed twice with Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS; Gibco) and 1% antibiotic–antimycotic (Gibco) and were centrifuged at 1300 rpm for 3 min. The SVF was filtered through a 100 μm strainer (BD Biosciences, San Jose, CA, USA). After centrifugation, the supernatant was discarded. The pellet was resuspended in culture medium, seeded in culture dishes and incubated overnight at 37 °C in a 5% CO2 atmosphere. Based on our hypothesis (Fig. 1A), the SVF was incubated, and after a certain period of time, the SF, which contains a large number of ASC that are initially adherent to the culture plate, and the VF, which contains a large number of endothelial cells floating in the supernatant, were isolated. The liquid was collected to obtain endothelial cells, which are nonadherent cells. More specifically, after 2 to 48 h, the culture supernatant of ASC was collected and filtered 1–2 times through a 10 ~ 40 μm pluriStrainer (PluriSelect Life Science, Leipzig, Germany). After centrifugation, the supernatant was discarded. The pellet was resuspended in endothelial cell medium (ECM; ScienCell, Carlsbad, CA, USA), a culture medium containing endothelial cell growth factor (ECGF; ScienCell), seeded in noncoated culture dishes and incubated overnight at 37 °C in a 5% CO2 atmosphere. After incubation for 24 h, the cells were washed 1–2 times with PBS, and the medium was then replaced with fresh medium. The resulting cell population was termed the processed HMVEC-A population to distinguish it from the population of HMVEC in the SVF from excised adipose tissue. HMVEC-A were maintained at 37 °C and 5% CO2 in medium containing ECGF. We isolated HMVEC-A by this method (Fig. 1B).
2.3 Magnetic activated cell sorting (MACS)
The isolated adipose-derived SVF was obtained after centrifugation, and the supernatant was discarded. The pellet was resuspended in culture medium and treated with 20 μl of FcR blocking reagent (to prevent nonspecific labeling of nonepithelial cells). The cells were magnetically labeled with anti-CD31 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) in the dark at 4 °C for 15 min and were then applied to the prepared LS column (Miltenyi Biotec, Bergisch Gladbach). Cluster of differentiation 31 (CD31)− cells were collected in the column flow-through, while CD31+ cells bound to the beads were flushed out by applying the plunger supplied with the column. Sorted CD31+ cells were plated into 100-mm-diameter plates and cultured in ECM containing ECGF (ScienCell). This experiment was independently repeated three times.
2.4 Fluorescence activated cell sorting (FACS)
The isolated adipose-derived SVF was obtained after centrifugation, and the supernatant was discarded. The pellet was resuspended in Hank’s balanced salt solution (Wisent Inc.) containing 2% FBS. The cells were incubated with monoclonal fluorescein isothiocyanate-conjugated antibodies specific for CD31 (PE; 555,446; BD Biosciences) for 30 min. The cells were subsequently washed with buffer, resuspended in 500 μl of cold buffer, and sorted in a MoFlo XDP cell sorter (Beckman Coulter, Indianapolis, IN, USA). This experiment was independently repeated three times.
2.5 Flow cytometric analysis
Passage 0 (P0) and passage 2 HMVEC-A, HUVEC and HMVEC were analyzed by flow cytometry. For flow cytometric analysis, cultured HMVEC-A, HUVEC, and HMVEC were trypsinized and resuspended in Hank’s balanced salt solution (Wisent Inc.) containing 2% FBS. The cells were incubated with monoclonal fluorescein isothiocyanate-conjugated antibodies specific for CD31 (PE; 555,446; BD Biosciences), CD146 (PE; 550,315; BD Biosciences), CD13 (PE; 555,394; BD Biosciences), and CD73 (PE; 550,272; BD Biosciences) for 30 min. The cells were subsequently washed three times with buffer, resuspended in 500 μl of cold buffer, and analyzed with a FACS Canto II flow cytometer (BD Biosciences, San Jose, CA). Unstained cells from each passage were used as controls. This experiment was independently repeated three times.
2.6 Western blotting
HMVEC-As isolated from the VF at different times were treated under appropriate conditions, washed with ice-cold PBS, and dissolved in lysis buffer. Total protein was extracted with T-PER Tissue Protein Extraction Reagent (Thermo Fisher Scientific, Rockford, IL, USA) containing 1% protease inhibitor cocktail (Sigma-Aldrich). The protein concentration was measured with a Quick Start Bradford protein assay kit (Bio-Rad Laboratories, Inc.). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis in a 10% gel and transferred to nitrocellulose membranes (Amersham, Buckinghamshire, United Kingdom). After blocking with 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 for one hour, the membranes were incubated with primary antibodies specific for CD31 (1:000, Abcam, Cambridge, UK, ab24590) and β-actin (1:5000, Sigma-Aldrich). Membranes were then incubated with a horseradish peroxidase-conjugated secondary antibody for two hours, and washed, and the antibodies were detected using a horseradish peroxidase substrate-enhanced chemiluminescence detection kit (GE Healthcare Life Sciences, Buckinghamshire, UK). The signals were quantified using a LAS 4000 image analyzer (Fujifilm, Tokyo, Japan) and Multi Gauge software (Fujifilm). Western blot analysis was performed in triplicate.
2.7 Proliferation assays
The proliferation of HMVEC-As isolated by three methods, Lonza HUVECs and Lonza HMVECs was assessed with a solution containing 10% Cell Counting Kit-8 reagent (Dojindo, Kumamoto, Japan). Cells were incubated in culture medium containing ECGF (ScienCell). The HMVEC-A, HUVEC, and HMVEC culture media were replaced with Cell Counting Kit-8 solution and incubated for two hours at 37 °C and 5% CO2. The supernatant was analyzed by enzyme-linked immunosorbent assay, and the absorbance was measured in a plate reader at 450 nm (SpectraMax PLUS384, Molecular Devices, Sunnyvale, CA, USA). The optical density of each well was calculated using the relative HMVEC-A standard curve. HMVEC-A proliferation was documented by imaging the cells under an inverted microscope (Olympus). The proliferation assay was conducted in triplicate.
2.8 In vitro tube formation assay
The endothelial tube formation assay was performed following the manufacturer’s protocol (BD Biosciences, Franklin Lakes, NJ, USA, https://www.bdbiosciences.com). Matrigel basement membrane matrix (Corning, Bedford, MA, USA) was thawed from − 20 °C by overnight incubation at 4 °C and was stored on ice until use. A 289 μl aliquot was dispensed into each well of a chilled 24-well culture plate using precooled pipette tips. The plate was incubated at 37 °C for 30–60 min to allow the Matrigel to solidify. The cell pellets were resuspended in ECM, and 300 μl of the cell suspension (containing approximately 1.2 × 105 HMVEC-A, HUVEC, or HMVEC) was added to each well containing Matrigel. The plate was incubated for 24 h at 37 °C and 5% CO2. Tube formation was visualized using an automated live-cell imager (LIONHEART™ FX, BioTek, Winooski, VT, USA). Analysis was performed with the provided software (Gen5™ 3.0, BioTek, Winooski, VT, USA). Tube formation was measured with ImageJ software.
2.9 DiI-Ac-LDL uptake assay
HMVEC-A, HUVEC and HMVEC were plated in 4-chamber slides at 2 × 104 cells/chamber. When the cells were 75% confluent, the culture medium was replaced with serum-free ECM for 24 h, and the cells were then incubated with 5 µg/ml 1,1′-dioctadecyl-1,3,3,3′,3′-tetramethylindocarbocyanine perchlorate acetylated LDL (DiI-Ac-LDL, Invitrogen Molecular Probes, Carlsbad, CA, USA) for four hours at 37 °C and 5% CO2. Then, the cells were washed, fixed with 4% paraformaldehyde at 4 °C for 30 min, and stained with 4′,6-diamidino-2-phenylindole (DAPI; VECTASHIELD® with DAPI) for 3 min. DiI-Ac-LDL uptake was assessed using fluorescence microscopy (Carl Zeiss, Axiovert 200, Oberkochen, Germany).
2.9.1 Immunohistochemistry
HMVEC-A (passage 2) were fixed with 4% paraformaldehyde (Sigma-Aldrich, Seelze, Germany), permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, USA) and blocked with PBS containing 10% FBS, 0.1% bovine serum albumin (BSA, Sigma-Aldrich) and 0.05% Tween 20 (Sigma-Aldrich). Cells were labeled with antibodies specific for the endothelial markers: CD31 (1:300 dilution; Abcam, ab24590) and VEGF (1:300 dilution; Abcam, ab46154) and were then incubated with the corresponding secondary antibodies (Chromeo™ 642 anti-mouse, 1:1000, ab60318, and Chromeo™ 488 anti-rabbit, 1:1000, ab60314; Abcam, UK). After incubation, cells were washed with PBS and mounted with medium containing DAPI (VECTASHIELD® with DAPI) to counterstain cell nuclei. Staining was assessed by fluorescence microscopy (Carl Zeiss, Axiovert 200).
2.9.2 Statistical analysis
Data are expressed as the mean ± SEM values. The significance of differences between groups was assessed by analysis of variance (ANOVA). All analyses were performed with GraphPad Prism version 5 (GraphPad Software, La Jolla, CA, USA). A p-value of < 0.05 was considered to indicate a statistically significant difference.
3 Results
3.1 Isolation of primary cells by the hypothesis that SVF derived from human adipose tissue is divided into an SF and a VF
The hypothesis of the SF and VF concept and the step-by-step procedure for isolating HMVEC-A from a single adipose tissue sample is shown in Fig. 1B. In brief, adipose tissue was harvested, minced, and digested with collagenase I for 30 min. The SVF was obtained by filtering the digest through a 100 μm cell strainer and was then seeded in a plate (1st culture plate). After 2–48 h of incubation, the supernatant VF was harvested and filtered through a 10–40 μm cell strainer for incubation of HMVEC-A on a plate (2nd culture plate). Human adipose-derived microvascular endothelial cells obtained by the VF isolation method were termed HMVEC-As. Therefore, two cell types—ASC and HMVEC-A—could be isolated from the same adipose tissue directly via the double harvesting system, which is an isolation process.
3.2 Analysis of the morphology and characteristics of the SF and VF separated by the method developed according to the hypothesis
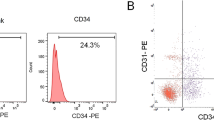
We were able to obtain HMVEC-A by isolating the VF from a single adipose tissue sample according to our hypothesis. Morphological analysis of each cultured cell type after separation of the SVF into the SF and VF indicated that ASC adhered to the bottom of the first culture plate and exhibited a spindle-shaped morphology, and HMVEC-As adhered to the second culture plate and exhibited a cobblestone morphology. In addition, in the analysis of DiI-Ac-LDL (an endothelial cells marker in both the SF and VF) uptake, Ac-LDL uptake could not be confirmed in the SF but was confirmed in the VF. In addition, FACS analysis confirmed the expression of the endothelial cell-specific markers CD31 and CD146. The percentages of cells expressing these markers were as follows: 53.1% for CD 31 and 44.3% for CD146 in the VF, and 13.3% for CD31 and 17.5% for CD146 in the SF. These results confirmed that the VF contained more endothelial cells (EC) than the SF (Fig. 2), indicating that this method is a reliable isolation method that allows the simultaneous isolation of EC and ASC.

Confirmation of EC characteristics of cells isolated from the SF and VF. Images of cells from the human adipose-derived stromal fraction (SF) and vascular fraction (VF) after seeding. Days 1, 3, and 7; magnification, 40 × . Cell nuclei were visualized by 4′6-diamidino-2-phenylindole (DAPI) (blue) staining. Uptake of DiI acetylated low-density lipoprotein (DiI-Ac-LDL: red) was confirmed. Day 3, magnification, 100 × . Cells in the SF and VF isolated from adipose tissue were labeled with an antibody specific for an endothelial cell (EC)-specific marker and analyzed by flow cytometry. Positivity for the endothelial-specific cell surface antigens CD31 (PECAM1) and CD146 (MCAM) was analyzed at passage 0 (n = 3)
3.3 Confirmation of medium specificity for the isolation of primary cells from the SF and VF by the method developed according to the hypothesis
The SVF was cultured for 24 h and then separated into SF and VF, and the specificity and suitability of ASC and EC medium for cell culturing were confirmed in each fraction. Cells isolated from the SF and VF were cultured with ASC medium (DMEM) and EC medium to confirm the difference in cells according to the medium. To this end, cells isolated from the SF and VF were cultured in DMEM (a stem cell medium) and EC Medium (an endothelial cells medium) independently and were then analyzed on the third day. This analysis confirmed that VF cells (HMVEC-A) cultured in DMEM exhibited a mixed cobblestone and spindle-shaped morphology and VF cells (HMVEC-A) cultured in EC Medium exhibited only a cobblestone morphology. In addition, SF cells (ASC) cultured in DMEM showed a distinct spindle shape and were confirmed to adhere to the culture plate and grow. Moreover, SF cells (ASC) cultured in EC Medium showed a mixed cobblestone- and spindle-shaped morphology (Fig. 3A). VF (HMVEC-A) and SF (ASC) cells cultured in each medium were confirmed by expression of the EC-specific marker CD31. The average percentage of CD31 + cells was 3.2% in the SF (ASC) + DMEM group, 5.8% in the SF (ASC) + EC Medium group, 23.1% in the VF (HMVEC-A) + DMEM group, and 39.6% in the VF (HMVEC-A) + EC Medium group. Significant differences were found between both the VF (HMVEC-A) + EC Medium and SF (ASC) + DMEM groups and the SF (ASC) + EC Medium and VF (HMVEC-A) + DMEM groups; significant differences were also seen between the SF (ASC) + DMEM and VF (HMVEC-A) + DMEM groups (*p < 0.05, **p < 0.01, ***p < 0.001) (Fig. 3B). In conclusion, VF (HMVEC-A) cells showed EC-specific characteristics when cultured in EC Medium, and EC Medium was found to be more suitable than DMEM. All analyses were performed three times at passage 0.

Separation of SF and VF after 24 h of SVF incubation followed by morphological analysis and FAS analysis according to the culture medium. A Morphology of cells isolated from the SF and VF on day 3 of culture in DMEM and EC Medium. The SF and VF were obtained from the same adipose tissue by the method developed according to our hypothesis. Magnification, 40 × . B CD31 marker expression as evaluated by FACS at P0 after incubation of SF and VF cells in DMEM and EC Medium. VF (HMVEC-A) + EC Medium increased CD31 expression compared to all groups (*p < 0.05, **p < 0.01, ***p < 0.001). In addition, CD31 expressions in the VF (HMVEC-A) + DMEM group increased (**p < 0.01) compared to the SF (ASC) + DMEM group
3.4 VF harvesting time for optimal HMVEC-A isolation
To find the optimal time for HMVEC-A isolation, the cell yield was assessed at each VF isolation time: 2 h (2H), 4 h (4H), 8 h (8H), 12 h (12H), 24 h (24H), and 48 h (48H). In addition, Western blot analysis was performed on VF harvested at each timepoint, and specific markers of ASC and EC were compared via FACS. When the yields were compared according to the VF harvest time, the yield was highest at 2 h, followed by 4 h and 12 h, the yield was similarly high, and the yield decreased in the order of 8 h, 24 h, and 48 h (Fig. 4A). To find the time when the expression of ASC-specific markers (CD13, CD73) was low and the expression of EC-specific markers (CD31, CD146) was significantly high in VF harvested at 2–48 h, we statistically compared EC-specific markers (CD31, CD146) with ASC-specific markers (CD13, CD73). The expression level was confirmed through FACS analysis. As a result of comparing the expression of ASC-specific markers (CD13, CD73) and EC-specific markers (CD31, CD146) at each harvest time of VF, the most significant difference was confirmed at 24 and 48 h (***p < 0.001) (Fig. 4B). Western blot analysis showed that CD31 expression peaked at 24 h (Fig. 4C). Based on these results, the optimal harvesting time was determined to be 24 h. However, there was no statistically significant difference. The VF harvesting time experiment was performed in triplicate.

Optimal VF harvesting time for HMVEC-A isolation, as determined by the yield, FACS analysis, and Western blot analysis. A The difference in yield from VF and SF harvested at 2–48 h of culture was confirmed. There was no statistically significant difference. B Expression level of ASC-specific markers (CD13 and CD73) and EC-specific markers (CD31 and CD146) were confirmed in VF harvested from 2-48 h incubation. As a result of analyzing each expression by comparing the ASC-specific marker and the EC-specific marker for each VF harvest time, a significant difference in expression of the EC-specific marker was confirmed at 24 and 48 h (***p < 0.001). C Quantitative differences in protein expression were confirmed by Western blot analysis of VF harvested at 2–48 h of culture. There was no statistically significant difference
3.5 Comparison of HMVEC-A isolation methods
The most widely used cell sorting methods, MACS and FACS, were compared with our method. After isolation and culture by three isolation methods—VF isolation, MACS, and FACS—EC characteristics were compared with those of HUVEC and HMVEC obtained from Lonza. Cells isolated by the VF isolation and MACS methods showed the most characteristic endothelial “cobblestone” morphology of EC. When the in vitro tube formation in Matrigel of HMVEC-A isolated by the three isolation methods was assessed, the results confirmed that the tube formation of HMVEC-A isolated by the VF isolation method was similar to that of the control HUVEC and HMVEC (Fig. 5A). In addition, the yield of the VF isolation method was significantly higher than that of the FACS isolation and MACS isolation methods (*p < 0.05, **p < 0.01). The doubling time was shorter for VF isolation and MACS isolation than for FACS isolation. As for the doubling time, VF isolation method was found to be the fastest in the order of VF isolation method, MACS isolation method and FACS isolation method, but there was no statistically significant difference. The average doubling times were 51.6 h for Lonza HUVEC, 39.1 h for Lonza HMVEC, 26.3 h for HMVEC-A isolated by VF isolation, 35.4 h for HMVEC-A isolated by MACS isolation and 70 h for HMVEC-A isolated by FACS isolation (Fig. 5B). Confirmation of the expression of the EC-specific markers CD31 and CD146 via FACS analysis revealed that among HMVEC-A isolated by the VF method, 48.7% expressed CD31 and 45.8% expressed CD146, and that among HMVEC-A isolated by MACS, 47.5% expressed CD31 and 38.2% expressed CD146. Additionally, among HMVEC-A isolated by FACS isolation, 24.1% expressed CD31 and 37.6% expressed CD146 (Fig. 5C). The percentages of cells expressing CD31 and CD146 were the highest for the VF isolation method. Lonza HUVEC and HMVEC were used as controls. The experiment was performed three times at passage 0.

Comparative analysis of properties according to the HMVEC-A isolation method. A Morphology and tube formation of HMVEC-A isolated by the vascular fraction (VF) isolation, magnetic-activated cell sorting (MACS) isolation method, and fluorescence-activated cell sorting (FACS) isolation methods were compared with Lonza HUVEC and HMVEC by phase-contrast microscopy; magnification, 40 × . B The VF isolation method produced a significantly higher yield than the FACS isolation method and MACS isolation method (*p < 0.05, **p < 0.01, n = 3). The doubling time was in the order of VF isolation method, MACS isolation method and FACS isolation method, and VF showed the fastest doubling time, but there was no statistically significant difference. The error bars indicate the standard errors of the mean of triplicate measurements. C FACS verification of EC-specific marker expression according to each isolation method. Positivity for the endothelial-specific cell surface antigens CD31 (PECAM1) and CD146 (MCAM) is indicated. Cells were analyzed at passage 2 (n = 3)
3.6 Characterization of HMVEC-As
After culture, the characteristics of HMVEC-A isolated from the adipose-derived SVF by the VF isolation method were confirmed by flow cytometry, immunocytochemistry (ICC) and in vitro tube formation assays. The morphology of passage 2 HMVEC-A was identical to the cobblestone-like morphology of EC (Fig. 6A), and uptake of DiI-Ac-LDL around the nucleus of the cell was confirmed (Fig. 6B). In addition, the expression of CD31 and VEGF was confirmed by immunocytochemistry (Fig. 6C). The angiogenic ability of HMVEC-A was established, and tube formation was confirmed to be maintained for 24 h (Fig. 6D). In addition, the results of flow cytometry indicated by the histogram showed that 82.6% of these cells were positive for CD31 and 77.3% were positive for CD146 (Fig. 6E). These results confirmed that HMVEC-A were successfully isolated.

Characterization of HMVEC-As. A Morphology of human microvascular endothelial cells (HMVEC-A) cultured by the vascular fraction (VF) isolation method. Magnification, × 40 × . B Uptake of DiI acetylated -low- density lipoprotein (DiI-Ac-LDL: red, DAPI: blue) was confirmed. Magnification, 100 × . C HMVEC-A were positive for the endothelial-specific cell surface markers CD31, and VEGF. Fluorescence microscopy showing positive immunoreactivity for CD31 (red) and VEGF (green). Magnification, 100 × . D The tube formation of HMVEC-A was maintained for 24 h. (40,000 viable cells/well were seeded in a 24-well polystyrene plate coated with Matrigel® Matrix (300 μl/well) and maintained in an incubator at 37 °C and 5% CO2). E Flow cytometric analysis of the HMVEC-A. HMVEC-A were positive for the endothelial-specific cell surface markers CD31, and CD146. The flow cytometric data represent 3 cell replicates with three experiments at the same passage
4 Discussion
The primary purpose of this study was to establish protocols and identify characteristics that facilitate the isolation of HMVEC-A from an adipose-derived SVF by simultaneously isolating and culturing microvascular endothelial cells and ASC from adipose tissue.
The cell population remaining in the supernatant discarded during the isolation and culture of ASC from an adipose-derived SVF is believed to include cells other than ASC. To this end, we hypothesized that SVF was separated into the SF, which is rich in ASC that are initially adherent to the culture plate but can be isolated, and the VF, which is rich in EC that are floating in the supernatant. Therefore, we collected and analyzed the discarded supernatant and confirmed the presence of EC. The time-course analysis to determine the optimal EC isolation time confirmed 24 h as the optimal isolation time. Thus, our hypothesis for simultaneously isolating EC and ASC in one sample was confirmed by the successful isolation of ASC and EC. Over the past few decades, EC has been isolated and cultured from various sources, and various studies have described the isolation of EC from human adipose tissue [23,24,25,26]. Adipose tissue can be used to study angiogenesis via the isolation and culture of large amounts of EC from a single sample. These cells are relatively easy to obtain, have a high proliferative capacity, and have been shown to be effective in therapeutic angiogenesis for ischemic diseases in animal models and clinical trials [27]. A high level of angiogenesis occurs in adipose tissue to provide a wide range of angiogenic capability necessary for growth [28, 29]. Thus, interest in the purification, cultivation, and molecular profiling of EC has recently increased, and a variety of isolation methods have been used [26,27,28]. For example, various methods, such as FACS, MACS, laser capture microdissection (LCM), and manual cell picking, can be used to isolate EC. However, these isolation methods have disadvantages such as the requirement for large amounts of material, dissociated cells, and high skill; nonspecific cell capture; contamination by neighboring cells; and low throughput [29,30,31]. However, our VF isolation method provides a much higher yield and shorter doubling time than other isolation methods, making HMVEC-A easier to obtain. Numerous studies have demonstrated the definite possibility of using angiogenic cells isolated from blood vessels derived from adipose tissue in regenerative medicine applications and have indicated the precise molecular mechanism of angiogenesis regulation in adipose tissue [32]. Many previous studies have used ASC to induce differentiation into vascular endothelial cells. These processes require repeated passages to extend growth to confluence, which is expensive because it is time-consuming and requires many reagents. Additionally, the potential for differentiation can be lost during long-term incubation in test tubes, the increased incubation time can reduce the possibility of angiogenesis, and the ability to repair damaged endothelium can be limited [33, 34]. However, based on our hypothesis, HMVEC-A isolated from adipose tissue showed the expression of EC fate markers without the induction of differentiation or repeated passaging, and their angiogenic ability was confirmed through in vitro tube formation assays. This method resulted in a higher yield with a shorter doubling time than other isolation methods; thus, HMVEC-A was easier to obtain by this method. The VF isolation method developed through our research was believed to allow various cells to be obtained from one sample. After collecting HMVEC-A at the optimal isolation time of 24 h by the VF isolation method, their morphological similarity to commercially available EC lines (HUVEC and HMVEC) was confirmed, and their vascularity was observed in vitro. These HMVEC-A exhibited marker expression and in vitro vascularity similar to those of the commercially available endothelial cell lines (HUVEC and HMVEC). This research strategy can thus be a promising method of developing cell sources for tissue regeneration and the treatment of defects, as it is safe and useful for creating autologous (self) grafts because of its ability to obtain several cells from a single tissue. Various tissue engineering strategies to improve angiogenesis, including both preangiogenic and preconditioned structures, have been proposed. EC has been cocultured with various types of cells, including MSC, ASC, and osteoblasts, and studies have been continuously conducted to enhance angiogenesis after in vivo transplantation [35, 36]. The majority of these studies have used commercially available HUVEC. However, if EC is stably maintained is are supplied from autologous tissues, they can be used as a basis for diverse studies in tissue engineering and regenerative medicine by integrating various growth factors with preconditioned tissue engineering structures or by changing the physical and chemical properties of scaffold materials. This adherence-based method, the VF isolation method, is a simple, reproducible, cost-effective technique that results in a high cell yield without the use of technically complex surface marker-dependent isolation methods. This VF isolation method is stable because it minimizes cell stress in a single isolation process and can allow simultaneous isolation of ASC and HMVEC-A from cells from autologous tissue to support a clinical application. Our study indicates that HMVEC-A can be isolated from adipose tissue while maintaining reproducibility. We successfully isolated and cultured HMVEC-A using a new protocol and then performed a detailed characterization of the isolated cells. When the most important characteristic of HMVEC-A—i.e., their vascularity—was evaluated, we found that the level of HMVEC-A tube formation was similar to that of Lonza HUVEC and HMVEC. These vascular endothelial cells were positive for the surface markers CD31 and CD146 and exhibited an angiogenic potential very similar to that of the Lonza HUVEC and HMVEC cell lines. Compared to the commonly used protocols, our protocol achieved an increased yield and shortened doubling time of EC in isolation and culture. Therefore, our protocol constitutes a simple and reliable technique for simultaneously isolating ASC and HMVEC-A from a single tissue according to the difference in adhesion between ASC (which adhere to the bottom of the culture plate) and HMVEC-A (which float in the culture supernatant), consistent with our hypothesis.
In conclusion, this study suggests that consistent with the new hypothesis, adipose tissue can be a source of new HMVEC-A as well as ASC and that a greater quantity of HMVEC-A can be obtained in a shorter amount of time than allowed by conventional isolation methods. We developed a protocol that allows easy and economically feasible isolation and culture of microvascular endothelial cells from adipose tissue without the need for complex processes. In this study, HMVEC-A, isolated at the same time as ASC from adipose tissue, expressed EC markers such as CD31, CD146, VEGFR2, and confirmed in vitro tube formation. This study reveals the potential of adipose tissue as source material for the isolation of HMVEC-A for tissue regeneration and its potential and possibilities, ranging from angiogenesis, organoid chip, disease modeling, drug screening, and clinical applications.
References
Marrella A, Lee TY, Lee DH, Karuthedom S, Syla D, Chawla A, et al. Engineering vascularized and innervated bone biomaterials for improved skeletal tissue regeneration. Mater Today (Kidlington). 2018;21:362–76.
Sarker MD, Naghieh S, Sharma NK, Chen X. 3D biofabrication of vascular networks for tissue regeneration: a report on recent advances. J Pharm Anal. 2018;8:277–96.
Rademakers T, Horvath JM, van Blitterswijk CA, LaPointe VLS. Oxygen and nutrient delivery in tissue engineering: approaches to graft vascularization. J Tissue Eng Regen Med. 2019;13:1815–29.
Mittal R, Woo FW, Castro CS, Cohen MA, Karanxha J, Mittal J, et al. Organ-on-chip models: implications in drug discovery and clinical applications. J Cell Physiol. 2019;234:8352–80.
Collins SD, Yuen G, Tu T, Budzinska MA, Spring K, Bryant K, et al. In vitro models of the liver: disease modeling, drug discovery and clinical applications. In: Tirnitz-Parker JEE, editor. Hepatocellular carcinoma. Brisbane (AU): Codon Publications. 2019. Chapter 3.
Olgasi C, Talmon M, Merlin S, Cucci A, Richaud-Patin Y, Ranaldo G, et al. Patient-specific iPSC-derived endothelial cells provide long-term phenotypic correction of hemophilia A. Stem Cell Reports. 2018;11:1391–406.
Williams IM, Wu JC. Generation of endothelial cells from human pluripotent stem cells. Arterioscler Thromb Vasc Biol. 2019;39:1317–29.
Sharath SS, Ramu J, Nair SV, Iyer S, Mony U, Rangasamy J. Human adipose tissue derivatives as a potent native biomaterial for tissue regenerative therapies. Tissue Eng Regen Med. 2020;17:123–40.
Chun SY, Lim JO, Lee EH, Han MH, Ha YS, Lee JN, et al. Preparation and characterization of human adipose tissue-derived extracellular matrix, growth factors, and stem cells: a concise review. Tissue Eng Regen Med. 2019;16:385–93.
Williams SK, Wang TF, Castrillo R, Jarrell BE. Liposuction-derived human fat used for vascular graft sodding contains endothelial cells and not mesothelial cells as the major cell type. J Vasc Surg. 1994;19:916–23.
Nishimura S, Manabe I, Nagasaki M, Hosoya Y, Yamashita H, Fujita H, et al. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes. 2007;56:1517–26.
Lee SJ, Lee CR, Kim KJ, Ryu YH, Kim E, Han YN, et al. Optimal condition of isolation from an adipose tissue-derived stromal vascular fraction for the development of automated systems. Tissue Eng Regen Med. 2020;17:203–8.
Nguyen A, Guo J, Banyard DA, Fadavi D, Toranto JD, Wirth GA, et al. Stromal vascular fraction: a regenerative reality? Part 1: current concepts and review of the literature. J Plast Reconstr Aesthet Surg. 2016;69:170–9.
Varma MJ, Breuls RG, Schouten TE, Jurgens WJ, Bontkes HJ, Schuurhuis GJ, et al. Phenotypical and functional characterization of freshly isolated adipose tissue-derived stem cells. Stem Cells Dev. 2007;16:91–104.
Dykstra JA, Facile T, Patrick RJ, Francis KR, Milanovich S, Weimer JM, et al. Concise review: fat and furious: harnessing the full potential of adipose-derived stromal vascular fraction. Stem Cells Transl Med. 2017;6:1096–108.
Ehrlund A, Acosta JR, Björk C, Hedén P, Douagi I, Arner P, et al. The cell-type specific transcriptome in human adipose tissue and influence of obesity on adipocyte progenitors. Sci Data. 2017;4:170164.
Ramakrishnan VM, Boyd NL. The adipose stromal vascular fraction as a complex cellular source for tissue engineering applications. Tissue Eng Part B Rev. 2018;24:289–99.
Bi H, Li H, Zhang C, Mao Y, Nie F, Xing Y, et al. Stromal vascular fraction promotes migration of fibroblasts and angiogenesis through regulation of extracellular matrix in the skin wound healing process. Stem Cell Res Ther. 2019;10:302.
Dobke M, Peterson DR, Mattern RH, Arm DM, Li WW. Microvascular tissue as a platform technology to modify the local microenvironment and influence the healing cascade. Regen Med. 2020;15:1313–28.
Kim JN, Lee JH, Oh DY, Yoo G, Jeon YJ, Moon SH, et al. Simple, and effective isolation and differentiation of endothelial cells and their progenitor cells from human fat tissue. Tissue Eng Regen Med. 2011;8:A37-43.
Ilesanmi AO. A novel way of harvesting a large population of endothelial cells for clinical and experimental use. Biomed Res Rev. 2018. https://doi.org/10.15761/BRR.1000120.
Schlie S, Gruene M, Dittmar H, Chichkov BN. Dynamics of cell attachment: adhesion time and force. Tissue Eng Part C Methods. 2012;18:688–96.
Sekiguchi H, Ii M, Jujo K, Yokoyama A, Hagiwara N, Asahara T. Improved culture-based isolation of differentiating endothelial progenitor cells from mouse bone marrow mononuclear cells. PLoS One. 2011;6:e28639.
Haynes BA, Huyck RW, James AJ, Carter ME, Gaafar OU, Day M, et al. Isolation, expansion, and adipogenic induction of CD34+CD31+ endothelial cells from human omental and subcutaneous adipose tissue. J Vis Exp. 2018;17:57804.
Van Pham P, Vu NB, Nguyen HT, Phan NK. Isolation of endothelial progenitor cells from human adipose tissue. BMRAT. 2016;3:645–52.
Bora P, Majumdar AS. Adipose tissue-derived stromal vascular fraction in regenerative medicine: a brief review on biology and translation. Stem Cell Res Ther. 2017;8:145.
Kalka C, Masuda H, Takahashi T, Kalka-Moll WM, Silver M, Kearney M, et al. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci. 2000;97:3422–7.
Pellegrinelli V, Carobbio S, Vidal-Puig A. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia. 2016;59:1075–88.
Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211–28.
Corvera S, Gealekman O. Adipose tissue angiogenesis: impact on obesity and type-2 diabetes. Biochim Biophys Acta. 2014;1842:463–72.
Fukumura D, Ushiyama A, Duda DG, Xu L, Tam J, Krishna V, et al. Paracrine regulation of angiogenesis and adipocyte differentiation during in vivo adipogenesis. Circ Res. 2003;93:e88-97.
Freiman A, Shandalov Y, Rozenfeld D, Shor E, Segal S, Ben-David D, et al. Adipose-derived endothelial and mesenchymal stem cells enhance vascular network formation on three-dimensional constructs in vitro. Stem Cell Res Ther. 2016;7:5.
Gharibi B, Hughes FJ. Effects of medium supplements on proliferation, differentiation potential, and in vitro expansion of mesenchymal stem cells. Stem Cells Transl Med. 2012;1:771–82.
Cribbs SK, Martin GS, Rojas M. Monitoring of endothelial dysfunction in critically ill patients: the role of endothelial progenitor cells. Curr Opin Crit Care. 2008;14:354–60.
Pill K, Melke J, Muhleder S, Pultar M, Rohringer S, Priglinger E, et al. Microvascular networks from endothelial cells and mesenchymal stromal cells from adipose tissue and bone marrow: a comparison. Front Bioeng Biotechnol. 2018;6:156.
Pill K, Hofmann S, Redl H, Holnthoner W. Vascularization mediated by mesenchymal stem cells from bone marrow and adipose tissue: a comparison. Cell Regen. 2015;4:8.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2017M3A9E2060428) and by a Ministry of Trade Industry and Energy of Korea (10063334).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
There are no animal experiments carried out for this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ryu, Y.H., Moon, SH., Kim, K.J. et al. A Novel Hypothesis and Characterization to Isolate Microvascular Endothelial Cells Simultaneously with Adipose-Derived Stem Cells from the Human Adipose-Derived Stromal Vascular Fraction. Tissue Eng Regen Med 18, 429–440 (2021). https://doi.org/10.1007/s13770-021-00332-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13770-021-00332-5