
Abstract
To evaluate the clinical and neuroimaging features of pediatric acquired demyelinating syndromes (ADS) in a tertiary pediatric neurology clinic in Turkey. All children diagnosed with any subset of ADS between 2013 and 2018 were included in this retrospective cohort study. Forty-two patients (21 female) with a median follow‐up period of 30 months were included. The median age of the patients at disease onset was 11 years (range 1.5–17 years). The most common pediatric ADS categories according to the International pediatric Multiple Sclerosis Study Group consensus classification criteria were acute disseminated encephalomyelitis (ADEM) and multiple sclerosis (MS), each of which seen in 15 patients, followed by clinically isolated syndrome (CIS) (n = 11) and Neuromyelitis Optica Spectrum Disorder (NMOSD) (n = 1). At the first clinical event, children with ADEM significantly differed from the children affected by MS and CIS in terms of the following parameters: median age at onset (7 vs. 13.5 and 14.5 years; p < 0.001), encephalopathy (93.3 vs 0% and 0%; p < 0.001), and basal ganglia/thalamus lesions (73.3 vs 9.1% and 9.1%; p < 0.001). The frequency of seizure and pleocytosis were higher in ADEM group than MS group (p < 0.05), whereas oligoclonal bands (p < 0.001) and periventricular white matter lesions (p < 0.01) were more frequently observed in MS patients. Rituximab was used with great success in the prevention of relapses in 3 patients: NMOSD (n = 1), MS (n = 1) and ADEM followed by recurrent optic neuritis (n = 1). Our results define the longitudinal disease course of various ADS categories in a single referral center. In addition, this study compares various clinical, laboratory and neuroimaging features between these ADS categories.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acquired demyelinating syndromes (ADS) is an umbrella term that encompasses a wide spectrum of inflammatory and demyelinating disorders of the central nervous system (CNS). ADS is exceedingly rare in children, with an estimated annual incidence of 0.6–1.66 cases per 100,000 children [1,2,3]. In 2012, several subcategories including multiple sclerosis (MS), monophasic acute disseminated encephalomyelitis (ADEM), multiphasic ADEM, neuromyelitis optica spectrum disorder (NMOSD) and clinically isolated syndrome (CIS) were addressed by the International Pediatric Multiple Sclerosis Study Group (IPMSSG) to improve consistency in terminology of clinical and basic researches [4].
The first clinical event of ADS is occasionally not adequate for accurate classification of a patient, requiring a period to describe whether it as a polyphasic or monophasic ADS. ADEM and CIS generally represent monophasic ADS, whereas MS, multiphasic ADEM and NMOSD have a polyphasic disease course. The involvement of one area (monofocal) or multiple areas (polyfocal) of the CNS, cranial imaging findings and the presence of autoantibodies further assist the diagnosis of the patient [5, 6]. Even at the initial clinical event early and accurate diagnostic identification of the relevant subcategory among these ADS categories is crucial since treatment options and prognosis vary substantially between ADS categories.
Pediatric MS with a disease onset before the age of 18 accounts for less than 10% of all MS cases [7]. Early recognition along with improving therapies have led to significant achievements in the quality of life and prognosis of MS patients. ADEM, which could affect patients of all ages, is the most frequently seen ADS of childhood and young adulthood. Encephalopathy and polyfocal neurologic dysfunction are the main manifestations of ADEM [8, 9]. Neuromyelitis optica spectrum disorders (NMOSDs) are characterized with optic neuritis (ON) and transverse myelitis (TM). The discovery of aquaporin-4 (AQP4) and MOG antibodies, which is also one of the supportive criteria for classification, has broadened our understanding of NMOSD [10]. Clinically isolated syndrome (CIS) is used to define a single and first demyelinating clinical episode with neurologic manifestations that last at least 24 h [4].
We have retrospectively evaluated consecutive children and adolescents diagnosed with ADS in a single referral center in Istanbul, to examine the clinical time course of various ADS categories and to compare clinical, laboratory and neuroimaging features, outcomes between different ADS categories.
Subjects and methods
This study was undertaken in a tertiary referral center (Cerrahpasa School of Medicine, Department of Pediatrics, Division of Pediatric Neurology) and all children diagnosed with any subset of ADS between January 2013 and 2018 were included in this retrospective longitudinal cohort study. All of the subjects had met the IPMSSG consensus classification criteria for pediatric MS and immune-mediated central nervous system demyelinating disorders including ADEM, NMOSD, CIS (including ON and TM) and pediatric ADEM as the first manifestation of MS and NMOSD [4]. Radiologic clinical syndrome (RIS) diagnoses of the patients were compatible with the modified diagnostic criteria proposed for RIS in children [11]. Clinical and laboratory data along with neuroimaging findings were obtained from patient records and hospital databases.
To exclude other inflammatory diseases, a standard diagnostic algorithm including comprehensive tests was implemented for all of the subjects. As routine clinical care, the patients are evaluated within one month after hospitalization period and followed up at 1–3-month intervals depending on the severity of the disease. All subjects were reclassified at the last follow-up visit according to IPMSSG guidelines [4]. Patients who have been diagnosed with ADEM or CIS at initial demyelinating event but experienced other episode(s) were re-evaluated whether or not they met MS or NMOSD.
The date of the first neurological symptom was considered as disease onset. The presenting manifestations were categorized into 8 groups: encephalopathy, seizures, sensory, motor, optic, brainstem, cerebellar and spinal symptoms. Preceding vaccination or infection, comorbid diseases, baseline clinical manifestations, medications and demographic data including age at disease onset and age at diagnosis, gender, duration of disease were retrieved from patient files. If available, laboratory data including viral serology, AQP4 antibodies, MOG antibodies, rheumatologic tests, cerebrospinal fluid (CSF) analysis, oligoclonal bands (OCB), immunoglobulin G (IgG) index were noted. All of the aforementioned data were also re-evaluated and noted at all of the demyelinating episodes after the initial event. Two or more oligoclonal IgG bands in CSF without corresponding bands in serum represented a positive test indicating intrathecal synthesis. An immunoglobulin G (IgG) index of more than 0.7 was considered positive.
The Expanded Disability Status Scale (EDSS) was used to assess disability. All imaging data from disease onset to the last visit were further reviewed by an experienced neuroradiology specialist (OK) who was blinded to the diagnosis and clinical history. Written informed consent was obtained from the legal guardians and the study protocol was approved by the institutional review board of the Cerrahpasa Medical School (333,206).
All statistical analyses were performed using SPSS statistical analysis software package version 20 (SPSS Inc., Chicago, IL). Categorical variables were presented using frequencies and percentages and continuous variables were summarized using median (minimum–maximum). Comparison of categorical variables between groups was performed using the chi-square or Fisher exact test (when appropriate). Non-parametric variables were compared by Kruskal–Wallis test between 3 independent groups. The significant threshold was set at p < 0.05. In case of significant difference between groups, pairwise comparisons were performed with post hoc Bonferroni correction.
Results
Demographic features
Overall, 42 patients (21 female) with a median follow‐up period of 30 months (range 9–60) were included in the study. The median age of the patients at disease onset was 11 years (range 1.5–17 years). At last visit, the most common pediatric ADS categories according to the IPMSSG consensus classification criteria were ADEM and MS, each of which was seen in 15 patients, followed by CIS seen in 11 patients. NMOSD was only in 1 patient and the age of disease onset was 13 years. Demographic and clinical characteristics of participants are listed in Table 1.
ADEM patients had a disease onset at an earlier age than the other ADS categories. The results showed that median age at onset varied among pediatric patients with ADS. Only one patient (6.6%) developed ADEM after 10 years of age (at age 14.5 years). Subjects with ADEM were significantly younger at the onset of first clinical CNS event than the MS group (7 vs. 14.5 years, respectively; p < 0.001).
Clinical data
There was significant difference in the frequency of patients with MS (n = 0) and ADEM (60%) who had reported an antecedent infection within 1 month before the first CNS clinical event (p < 0.001). None of the subjects with ADS reported a vaccination within the last month before the onset of disease (Table 1).
The frequency of high fever preceding the neurologic symptoms was comparable in patients with MS and CIS (0 vs 9.1%), but it was significantly higher in patients with ADEM (60%, (p < 0.001). In ADEM patients, the incidence of seizure and encephalopathy at the time of disease onset were 33.3 and 93.3%, respectively. Neither seizure nor encephalopathy was observed among patients with MS and CIS.
At the first clinical CNS event, motor paralysis was observed more frequently in patients with ADEM than in patients with MS (66.7 vs 33.3%, respectively), but it was not significant in Bonferroni post hoc correction (p = 0.204). We observed a higher frequency of sensory disturbance in patients with MS and CIS (33.3 and 36.4%, respectively) than in patients with ADEM (0%). However, after Bonferroni post hoc correction, only the difference between ADEM and CIS was found as significant (p = 0.022).
A 16-year-old girl was eventually diagnosed with MS after 2 years of follow-up with RIS. Initial cranial MRI, which was ordered to evaluate headache, demonstrated MS-like plaques. Follow-up MRIs showed contrast enhancement in addition to the previous plaques. Increased IgG index (1.28) level and presence of OCB were suggestive of MS. A diagnosis of MS was also secured clinically with the emergence of diplopia after 2 years of duration without any therapy.
Laboratory data
Laboratory data are presented in Table 2. A higher percentage of CSF pleocytosis was detected in children with ADEM than MS patients (62 vs 8%). Elevated IgG index was encountered in 78% of the MS patients, higher than the 25 and 17% of the ADEM and CIS group, respectively. After Bonferroni post hoc correction, MS patients had a higher rate of oligoclonal bands in CSF compared to the ADEM group (93 vs 11%, respectively; p < 0.001).
Both IgG index and oligoclonal bands were positive in 7 out of 15 MS subjects. Of the 24 ADS patients, who were tested for MOG antibodies, 3 were positive. MOG antibodies were detected in only ADEM group, with a frequency of 27.2% (n = 3/11). One out of 3 anti-MOG seropositive ADEM patients experienced 3 episodes of ON after the initial ADEM episode (ADEM-ON). Of the patients with anti-MOG antibodies, one patient has never experienced an episode during 3 years of follow-up. In two patients, the initial ADEM event at 10 years of age was followed by one or several events of ON. One patient had three ON episodes within 3 years, with an interval of one year. There was no episode of ON within 1.5 years of follow-up after administration of rituximab. Likewise, ON developed in another patient at the end of the second month after the initial ADEM event. Only one patient met all the criteria for NMOSD with a positive AQP4 antibody result and this patient was negative for anti-MOG antibody.
Neuroimaging findings
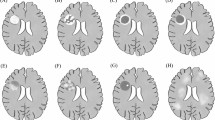
During the first clinical CNS event, the median numbers of cranial MRI findings are given in Table 2. Basal ganglia/thalamus lesions were detected in 73% of subjects with ADEM and 9.1% with MS and CIS (p < 0.001). After Bonferroni post hoc correction, periventricular white matter lesions were detected more frequently in MS group than in patients with ADEM (80 vs 26.7%, respectively; p = 0.009). There was no significant difference between patients with ADEM and MS with respect to the frequency of optic nerve lesion (6.7 vs 13.3%, respectively). After Bonferroni post hoc correction, statistically significant difference in gadolinium-enhancing lesions was documented between patients with MS (73%) and CIS (18%) (p = 0.015). Periventricular white matter lesions were detected more frequently in MS group than in patients with ADEM (80 vs 26.7%, respectively; p = 0.003). In ADEM group, all patients except one had cerebral MRI lesions (93.3%), and 3 patients (20%) had solely cerebral lesions. The common features of our 3 MOG-Ab-positive ADEM and ADEM-ON cases on MRI were subcortical, cortical, basal ganglion and thalamic involvements at the first clinical event.
Treatment, adverse events and outcome
In the induction therapy period, most patients with pediatric ADS received immunosuppressive therapy, including intravenous high-dose methylprednisolone, oral corticosteroids, and intravenous immunoglobulin (IVIG). Interferon beta was used in the remission phase in 13 patients with MS (86.6%). Glatiramer acetate was initiated in a patient who experienced an initial clinical CNS event at 17 years of age with high number of lesions. Interferon beta treatment was switched to glatiramer acetate due to elevated transaminases during treatment in another patient. New episode of CNS demyelination occurred in 2 patients, while they were on interferon beta treatment. Natalizumab was started in one of the two patients who had a new attack during interferon treatment, and rituximab was started in the other. Of 4 MS patients with neurological sequelae, 2 patients had mild motor disturbance, 1 had sensory disturbance and 1 had ophthalmoplegia, all of which were as a result of clinical episodes before therapy.
Thirteen out of 15 patients with ADEM, were treated with intravenous high-dose methylprednisolone followed by oral corticosteroids (for 2 weeks, then gradually tapered and withdrawn within the next 2 weeks) along with symptomatic therapy. Six patients received both the standard corticosteroid regime and IVIG therapy. Plasma exchange was performed in a patient who was unresponsive to this combination therapy. Given 3 episodes of ON within 3 years and positive anti-MOG antibodies, rituximab was initiated with a great success in the prevention of relapses. Fifteen ADEM patients except one (93.3%) completely recovered after the initial CNS clinical event. The neurological sequel in one patient (7%) was a moderate motor disturbance (weakness in left arm).
Eight of 10 patients (80%) with pediatric CIS were treated with high-dose IV methylprednisolone. In addition, oral corticosteroid was used in 6 patients with ON for 4 weeks. Despite the IVIG therapy, a mild motor disturbance as a sequela was observed in a patient who had been also unresponsive to high-dose intravenous methylprednisolone. In another patient due to the absence of any improvement in visual acuity (4/10) was considered as a neurological sequela.
The initial clinical event of the NMOSD patient was bilateral visual loss. High-dose intravenous methylprednisolone therapy failed to induce a favorable recovery in the left eye. The episodes including visual, motor and sensorial impairment were observed even after initiation of azathioprine. Plasma exchange prevented further relapses; however, complete control of relapses was only achieved by rituximab with 2 years of attack-free period. Visual loss in left eye (visual acuity 1/10) was considered as a neurological sequela after the therapy.
Discussion
In the present study, we have assessed the clinical characteristics of various ADS categories according to IPMSSG definitions and compared clinical, laboratory and neuroimaging features between these ADS categories. Demographic features of our pediatric ADS group were similar to those reported in previous studies, but there were some unique differences. The female/male ratio in ADS patients was 1:1. Although a previous Turkish and European study report a female: male ratio of approximately 1.8:1 and 2.8:1, respectively, in our MS cohort, males outnumbered females 1–0.7 [12, 13].
The frequency of ADEM and MS in our study group was equal, both of which were seen in 15 patients. We have confirmed that the median age of disease onset was younger in ADEM than that in MS. The median age at disease onset for MS and ADEM in our study was in line with those reported by an Italian prospective study (mean age: 14.4 and 8.1, respectively) [14], nationwide survey conducted in Germany [15] and a Turkish multicenter study [12]. In a Japanese study, the mean age at disease onset for MS, CIS and ADEM were found as 8.3, 6.2, 5.5 years, respectively, all of which were younger than the previous reports [16]. The authors proposed that genetic or environmental factors could be associated with these younger disease onsets.
Preceding viral and bacterial infections may induce the development of ADS, particularly ADEM [17,18,19]. In our study, a history of antecedent infection was reported by 9 patients with ADEM, in only 2 of whom specific infectious agents have been identified. The serologically proven infectious agent was EBV in one ADEM patient. However, the other patient experienced the first clinical ADEM episode during the skin rashes related to varicella. Interestingly, the ADEM episode and skin rashes observed at the same time period in our case. Since there are a considerable number of reports documenting significant benefits from anti-infectious therapy in ADS, detailed serological and microbiological evidence of infectious agents should be investigated [18].
In accordance with the literature, the most common clinical finding in our ADEM patients was motor impairment [9, 17, 20]. Pediatric MS can result in a broad spectrum of clinical features. In agreement with earlier data, the most frequent manifestations of our MS group were motor, brainstem and sensory impairment [12, 21]. The clinical phenotype of ADEM is characterized by polyfocal neurological deficits and the presence of encephalopathy is a prerequisite for the IPMSSG definition; however, in MS, encephalopathy never accompanies the episodes [4]. Encephalopathy was observed in 93.3% of our patients with ADEM, whereas none of the patients in MS group had encephalopathy. Fever, systemic illnesses and postictal symptoms, all of which are occasionally misdiagnosed as encephalopathy, are more frequent at younger ages.
Seizure in first clinical CNS episode was noted in 33.3% of the patients with ADEM. However, neither MS patients nor CIS patients had seizure during first clinical event. Likewise, in two different Turkish studies the frequency of seizure in ADEM and MS patients were found as 26.7 and 5.5%, respectively [22, 23]. Among patients with pediatric MS, seizure was observed with a frequency of 3% and 0% in a Dutch and USA study, respectively [24, 25]. Compared to the published reports, a Japanese study reported higher rates of seizure (29%) in pediatric MS patients at the time of disease onset [16]. The pathogenesis of seizures and the basis of its association with MS are still unknown. Cortical lesions with surrounding inflammation and edema have been postulated as a possible explanation for increased seizure risk in MS by disrupting the electrophysiological activity [26]. On the contrary, seizure was not observed in any of the 4 MS patients with cortical lesions. Of the 6 ADEM patients with seizures, 3 (50%) had cortical lesions on MRI. The difference in the frequency of seizures between MS and ADEM may be explained by the higher level of inflammatory response in ADEM than in MS.
MOG-Ab-associated disease presenting with grey matter lesions and seizures has been increasingly recognized in children [27]. Observation of isolated seizures without clinical features of ADEM, during the initial episode of MOG-Ab-associated demyelination suggested a causal link between MOG antibodies and autoimmune epilepsy [28]. Higher rates of relapses and seizures in ADEM patients with positive MOG-antibodies were found in a study, indicating a childhood immune-mediated chronic disease with seizures [29]. On the contrary, seizure was never noted either during episodes or after post-ADEM period of our three ADEM patients with anti-MOG antibodies. Future prospective studies with large patient numbers are needed to replicate the association of MOG antibodies and isolated seizures.
Interferon-beta (IFN-β) or glatiramer acetate (GA) has been recommended as a first-line therapy for pediatric MS by IPMSSG [30]. Thirteen patients (87%) in our MS group received IFN-β as a first-line therapy. Of the 3 patients with adverse event, one patient switched to GA therapy due to elevated transaminases. Natalizumab treatment was started and resulted in disease inactivity over 2 years of follow-up in another patient. Lower annual relapse rates have been observed in adult MS patients with natalizumab therapy; however, there are concerns regarding increased risk of progressive multifocal leukoencephalopathy (PML) [31]. There has been no PML case associated with natalizumab therapy in pediatric MS patients. After a median of 34 months with natalizumab therapy, 58% percent of 101 pediatric MS patients with aggressive clinical course were free of activity both clinically and radiologically at last observation [32].
There has been substantial progress regarding pediatric ADS over the past decades. Periodic updates and a better description of the IPMSSG definitions regarding ADS and subcategories, discovery of novel biomarkers of neuroinflammation have significantly facilitated the differential diagnosis of ADS, thereby leading to early initiation of respective therapies.
References
Banwell B, Kennedy J, Sadovnick D et al (2009) Incidence of acquired demyelination of the CNS in Canadian children. Neurology 72:232–239
Ketelslegers IA, Catsman-Berrevoets CE, Neuteboom RF et al (2012) Incidence of acquired demyelinating syndromes of the CNS in Dutch children: a nationwide study. J Neurol 259:1929–1935
Langer-Gould A, Zhang JL, Chung J et al (2011) Incidence of acquired CNS demyelinating syndromes in a multiethnic cohort of children. Neurology 77:1143–1148
Krupp LB, Tardieu M, Amato MP et al (2013) International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler 19:1261–1267
Waldman A, Ghezzi A, Bar-Or A et al (2014) Multiple sclerosis in children: an update on clinical diagnosis, therapeutic strategies, and research. Lancet Neurol 13:936–948
Banwell B, Bar-Or A, Arnold DL et al (2011) Clinical, environmental, and genetic determinants of multiple sclerosis in children with acute demyelination: a prospective national cohort study. Lancet Neurol 10:436–445
Ness JM, Chabas D, Sadovnick AD et al (2007) Clinical features of children and adolescents with multiple sclerosis. Neurology 68:S37-45
Tenembaum S, Chitnis T, Ness J et al (2007) Acute disseminated encephalomyelitis. Neurology 68:S23-36
Schwarz S, Mohr A, Knauth M et al (2001) Acute disseminated encephalomyelitis: a follow-up study of 40 adult patients. Neurology 56:1313–1318
Wingerchuk DM, Banwell B, Bennett JL et al (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85:177–189
Makhani N, Lebrun C, Siva A et al (2017) Radiologically isolated syndrome in children. Neurol Neuroimmunol Neuroinflamm 4:e395
Yılmaz Ü, Anlar B, Gücüyener K et al (2017) Characteristics of pediatric multiple sclerosis: the Turkish pediatric multiple sclerosis database. Eur J Paediatr Neurol 21:864–872
Simone IL, Carrara D, Tortorella C et al (2002) Course and prognosis in early-onset MS: comparison with adult-onset forms. Neurology 59:1922–1928
Atzori M, Battistella PA, Perini P et al (2009) Clinical and diagnostic aspects of multiple sclerosis and acute monophasic encephalomyelitis in pediatric patients: a single centre prospective study. Mult Scler 15:363–370
Pohl D, Hennemuth I, Von Kries R, Hanefeld F (2007) Paediatric multiple sclerosis and acute disseminated encephalomyelitis in Germany: results of a nationwide survey. Eur J Pediatr 166:405–412
Yamaguchi Y, Torisu H, Kira R et al (2016) A nationwide survey of pediatric acquired demyelinating syndromes in Japan. Neurology 87:2006–2015
Dale RC, de Sousa C, Chong WK et al (2000) Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain 123(Pt 12):2407–2422
Özkale Y, Özkale M, Demir E, Alehan F (2012) Acute disseminated encephalomyelitis associated with Influenza A H1N1 infection. Pediatr Neurol 47:62–64
Stonehouse M, Gupte G, Wassmer E, Whitehouse WP (2003) Acute disseminated encephalomyelitis: recognition in the hands of general paediatricians. Arch Dis Child 88:122–124
Gudbjornsson BT, Haraldsson Á, Einarsdóttir H, Thorarensen Ó (2015) Nationwide incidence of acquired central nervous system demyelination in Icelandic children. Pediatr Neurol 53:503–507
Öztürk Z, Yllmaz Ü, Konuśkan B et al (2018) Multiple sclerosis with onset younger than 10 years in Turkey. Neuropediatrics 49:51–58
Erol I, Özkale Y, Alkan Ö, Alehan F (2013) Acute disseminated encephalomyelitis in children and adolescents: a single center experience. Pediatr Neurol 49:266–273
Durmus H, Kurtuncu M, Tuzun E et al (2013) Comparative clinical characteristics of early- and adult-onset multiple sclerosis patients with seizures. Acta Neurol Belg 113:421–426
Alper G, Heyman R, Wang L (2009) Multiple sclerosis and acute disseminated encephalomyelitis diagnosed in children after long-term follow-up: comparison of presenting features. Dev Med Child Neurol 51:480–486
Neuteboom RF, Boon M, Catsman Berrevoets CE et al (2008) Prognostic factors after a first attack of inflammatory CNS demyelination in children. Neurology 71:967–973
Sponsler JL, Kendrick-Adey AC (2011) Seizures as a manifestation of multiple sclerosis. Epileptic Disord 13:401–410
Hacohen Y, Wong YY, Lechner C et al (2018) Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody–Associated disease. JAMA Neurol 75:478–487
Ramanathan S, O’grady GL, Malone S, et al (2019) Isolated seizures during the first episode of relapsing myelin oligodendrocyte glycoprotein antibody-associated demyelination in children. Dev Med Child Neurol 61:610–614
Rossor T, Benetou C, Wright S et al (2020) Early predictors of epilepsy and subsequent relapse in children with acute disseminated encephalomyelitis. Mult Scler 26:333–342
Chitnis T, Tenembaum S, Banwell B et al (2012) Consensus statement: evaluation of new and existing therapeutics for pediatric multiple sclerosis. Mult Scler 18:116–127
Polman CH, O’Connor PW, Havrdova E et al (2006) A randomized, placebo-controlled trial of Natalizumab for relapsing multiple sclerosis. N Engl J Med 354:899–910
Ghezzi A, Moiola L, Pozzilli C et al (2015) Natalizumab in the pediatric MS population: results of the Italian registry. BMC Neurol 15:174
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Ethical approval
The study received approval from the Ethics Committee of the Istanbul University-Cerrahpasa, Cerrahpasa School of Medicine.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kilic, H., Mavi, D., Yalcinkaya, B.C. et al. Evaluation of inflammatory acquired demyelinating syndromes in children: a single-center experience. Acta Neurol Belg 122, 1485–1491 (2022). https://doi.org/10.1007/s13760-021-01703-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-021-01703-4