
Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease leading to motor neuron damage. In this study, the clinical, demographic, and genetic features of ALS patients in the city of Sakarya, Turkey, were investigated. Patients with an established diagnosis of ALS according to the Awaji criteria were included. Age, sex, age at onset of ALS, initial complaints, consanguineous marriage, and genetic features were retrospectively investigated. Conventional genetic analysis and NGS were used for molecular evaluation of patients. A total of 55 probands (10 familial, 45 sporadic) in whom ALS was suspected due to their phenotypic features were included. Thirty-two patients were male (58.2%), and 23 were female (41.8%); their mean ages were 62.65 ± 13 years. The mean age of onset for 37 familial patients from 10 families was 49.9 years. Two cases had juvenile-onset. Fourteen (25.5%) bulbar-onset versus 40 (72.7%) limb-onset patients were detected; one patient had both. Six (10.9%) patients showed marked frontotemporal dementia. Twenty-nine (52.7%) patients died during the follow-up period. Genetic analysis identified causative variants in eleven cases, carrying variants in six different ALS genes (C9orf72, SOD1, VCP, SPG11, TBK1, and SH3TC2). Genetic investigations have revealed more than 40 genes to be involved in the pathogenesis of ALS. Our relatively small study cohort restricted to one province of Turkey, however, prone to migration, consists of 10/55 familial ALS cases, which harbor two rare (SH3TC2-p.Met523Thr and TBK1-p.Glu643del) and two novel (SPG11-p.Lys656Valfs*11 and VCP-p.Arg191Pro) mutations contributing to the literature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects motor neurons in the cortex, brain stem, and spinal cord. The prevalence of ALS ranged from 1.07–11.31 per 100.000 in meta-analysis of twenty-six different studies [1]. Most of study suggested a slightly increasing prevalence of ALS over years [2, 3]. The prevalence of ALS appears to vary between ethnic groups. The prevalence of European American ALS is higher than African American ALS patients (5.4:2.3) [4]. A study reported that the prevalence was 7.3 per 100.000 in Thrace region of Turkey [5].
Chiò et al. reported that the incidence rate was 2, Ryan et al. reported 3.1 per 100.000 individuals [6, 7]. When the disease was evaluated according to gender, male sex has since long being considered a risk factor for ALS. The studies reported that male-to-female ratios in ALS between 1.2 and 1.5 [8, 9]. If no family history is identified, the diagnosis is assumed to be sporadic. The sporadic ALS comprises 67% males [10]. In familial ALS, ratio of males to females is 1:1 [10]. A study reported that the mean male-specific annual incidence was 1.8, and female-specific was 1.3 per 100.000/year [7]. A prospective population-based parent–offspring heritability study was reported that the annual age-standardized and sex-standardized incidence of ALS did not change with time [7]. Most studies detected that the incidence of sporadic was 1–2 per 1000.000 individuals, years ALS in the Western countries [11,12,13].
ALS has a complex genetic origin. Heritability measures the extent to which genetic variation between individuals. Family history of ALS in a first degree relatives increases the risk of developing ALS 1.4-fold.[7]. A study reported that if there was no family history of ALS, the mean lifetime heritability was 36.9% [7].
ALS increases exponentially with age. Sporadic ALS usually occurs in mid-to-late 50 s [10]. The age of onset for sporadic ALS is 58–63 [14, 15]. A peak incidence in the age group 70–74 years [16]. About 10–15% of the disease has a familial ALS form occurs in patients in their late teens or early adulthood [10]. A study reported that affected offspring were younger than their parents in assessment using parent–child ALS dyads [7].
ALS patients have a more limited survival after diagnosis. A slow form of ALS with a survival of 10 years or longer is seen in 10% of the patients.
The pathophysiology of ALS is characterized by the loss of pyramidal Betz cells (motor cortex) as well as by loss and degeneration of the large anterior horn cells (spinal cord and lower cranial motor nuclei of the brainstem) [17]. Damaged motor neurons display intracellular aggregates that form distinct ubiquitinated inclusions, which play an important role in the pathology of the disease. There is no single or specific test for ALS diagnosis. Diagnosis is established by proving the presence of a progressive disorder with manifestations of typical upper and lower motor neuron involvement and exclusion of other etiologies by clinical examination and electromyography [13, 18, 19]. The Awaji criteria are used for diagnosis and classification [20]. The Awaji criteria recommended that neurophysiological data should be used in the context of clinical information. In addition, fasciculation potentials associated with signs of reinnervation were considered as evidence of lower motor neuron lesion, in particular in cranial-innervated or strong limb muscles.
Over the last decade, genetic discoveries had a profound impact on our understanding of ALS and there was an exponential growth in the phenotypes labeled as ALS. ALS is now considered not as a single disease, but rather as a syndrome, consisting of diseases due to a series of non-overlapping mechanisms, which, however, all culminate in motor neuron death. Genetics has provided novel insights into the pathogenesis of ALS, yet also uncovered novel layers of complexity, increasingly influencing diagnostics and treatment. Today, more than 40 genes are implicated in the pathogenesis of ALS, which can be summed up in three main groups as disrupted protein homeostasis, impaired RNA metabolism, and axonal transport defects [21]. Protein aggregates are a hallmark of several neurodegenerative diseases. Thus, misfolded SOD1 aggregates and other defective genes’ products like UBQLN2, VCP, OPTN, and TBK1, involved in ubiquitin-proteosome system, ER-mediated degradation, and autophagy have been the main focus of ALS research. However, identification of mutations in RNA-binding proteins TDP-43 and FUS has associated the defects in RNA processing with ALS pathology. TDP-43 protein inclusions are observed in motor neurons of all ALS cases, with the exception SOD1 and FUS-based ALS. In case of ALS, TDP-43 and FUS, normally located in the nucleus, are exported to cytoplasm and cause formation of stress granules, which sequester several RNA-binding proteins, silencing mRNA translation. Moreover, disrupted RNA metabolism in ALS is further supported by the formation of nuclear and cytoplasmic RNA foci that harbor RNA–protein aggregates as a result of the dynamic hexanucleotide repeat expansions in the promoter region of C9orf72, which is the most frequent genomic variation both in fALS and in sALS worldwide. Finally, altered cytoskeletal dynamics, due to pathogenic variations in several proteins involved in axonal integrity and transport (e.g., PFN1, DCTN1, NEFH and TUBA4A), is the third main pathology underlying ALS.
Most ALS cases are sporadic, with the exception of 10% showing hereditary patterns [21, 22]. Sporadic ALS (sALS) is considered to be a result of the involvement of genetic and environmental factors; its etiology is not yet known. Familial ALS, although low in percentage, is a model to study sALS, since the pathological signs of the familial and sporadic disease exhibit great similarity. However, even fALS is phenotypically and genetically heterogeneous. In the majority of fALS cases, an autosomal dominant inheritance pattern has been described, although recessive and X-linked forms also exist [23]. In addition, the Mendelian inheritance of ALS indicates that the genetic defects expressed become selectively toxic, indicating incomplete penetrance. This, in turn, suggests a significant role for genetic factors in the sporadic form of ALS. Twelve percent of fALS cases are explained with SOD1 mutations, 4–5% with FUS and TARDBP each, and 40% with C9orf72 [24,25,26,27]. These are followed by ANG, OPTN, UBQLN2, and VCP [21, 23]. These same gene mutations explain approximately 12% of sporadic cases.
Clinical and pathological findings of fALS and sALS cases are indistinguishable, with the exception of the age at onset being several years earlier in fALS. Genetic transmission is usually autosomal dominant with various penetrance ratios in fALS [20, 28]. To predict the clinical course and prognosis of ALS, genotyping is important. Next-generation sequencing (NGS) has made a great impact on our progress toward the genetic diagnosis of neurological disorders. Especially whole exome sequencing (WES) has been applied both in research and in diagnostic settings, using single cases or case–control studies with thousands of subjects to identify novel disease-causing genes. Currently, the diagnostic success rate of WES is approximately 40%, which can be higher in familial cases and in consanguineous families [29]. In this study, we investigated the phenotypic and genotypic features of ALS cases in Sakarya, Turkey, using conventional and next-generation sequencing methods.
Materials and methods
We reviewed the clinical findings and genetic analyses of 55 patients, who were diagnosed with ALS in our center between June 2012 and March 2019. Brain and spinal MRI and electrophysiological tests were performed in all patients to diagnose the disease. The patients’ initial symptoms (bulbar, upper limb, lower limb-onset) were recorded. Physical examination findings, including a detailed neurological examination, dysmorphic features, and course of the disease were evaluated. Further, consanguineous marriage and the presence of similar diseases within the family were recorded and clinical phenotypes were categorized. Pedigree analysis pointed to genetic transmission in several cases. For a definitive diagnosis samples were subjected to genetic analyses and peripheral venous blood samples obtained from patients and family members were investigated. Genomic DNA was extracted from whole blood using the MagNAPure Compact DNA Isolation System (Roche, Switzerland). Conventional screening of the most common ALS genes (SOD1, TARDBP (exon 5), FUS (exons 14 and 15) and UBQLN2) was performed using polymerase chain reaction (PCR) followed by Sanger sequencing (Macrogen, Korea). The hexanucleotide repeat expansion in the promoter region of the C9orf72 genewas tested using repeat-primed PCR and fragment length analysis. WES coupled with bioinformatic analysis was performed in a subset of unsolved cases. Segregation analysis for the candidate variants was performed in available family members. The patients with spinobulbar muscular atrophy were excluded. Twenty-five sALS samples were subjected to whole genome sequencing (WGS) in the framework of Project MinE.
Results
The demographic features of 55 ALS patients included in this study are compiled in Table 1. The patients were divided into three groups according to Awaji criteria: definite ALS (74.5%), probable ALS (18.2%), and possible ALS (7.3%).
Except for two cases with juvenile-onset, the mean age of onset of the patients was 60.75 ± 10.25 years, and the mean age at the time of onset of disease complaints was 57.94 ± 14.41 years. In the two juvenile cases, complaints started at the ages of 17 and 20. The study included 37 fALS patients from 10 (18.2%) families and 45 (81.8%) sALS cases (Table 1). The mean age at onset was 49.9 (17–77) years for fALS (Table 2). Two (3.6%) sALS patients were 80 years or older at disease onset. Fourteen (25.5%) bulbar-onset ALS patients were detected and all of them were sALS. Among 40 (72.7%) patients with limb-onset disease, 18 had lower (13 sALS, 5 fALS), 21 had upper (16 sALS, 5 fALS) extremity symptoms, and one patient showed both. Only one patient in our study cohort had bulbar and upper extremity symptoms at the same time, as an initial symptom of ALS (Table 1). A patient with bulbar-onset had weakness of neck flexor muscles as a rare symptom of ALS. The patient had additional findings including thenar–hypothenar atrophy, hyperactive deep tendon reflexes, and weakness three months later. Myasthenia graves were excluded by negative anti-acetylcholine receptor antibody, no response to edrophonium test, no abnormality in repetitive nerve stimulation, and single-fibre electromyography. He had normal value of creatinine kinase. Clinical and control electrophysiological study confirmed ALS diagnosis after three months.
Muscle atrophy was observed in all subjects, but the distribution of the atrophy was wide in four patients. These patients had bilateral flail arm syndrome also known as ‘man-in-barrel syndrome’ in clinical follow-up after the diagnosis. Six (10.9%) ALS patients showed overtly frontotemporal dementia (FTD) (4 sALS, 2 fALS).
During the follow-up period, twenty-nine (52.7%) patients died. Most of these patients deceased from respiratory failure, the cause of death could not be determined in some other patients. The diaphragm pacing system was applied to two patients. One patient lived for 3 years after application of the diaphragm pacing system, another patient had diaphragm pacing system for a 1 year.
Genetic analysis identified causative variants in 20% of cases in the cohort under study (11/55) (Table 2). The C9orf72 hexanucleotide repeat expansion was detected in six ALS cases (10.9%). Four of these cases had fALS and two were sporadic. Three female and three male patients were recorded. The mean age of disease onset of C9orf72 expansion carriers was 57 (53–62) years. One of them had bulbar-onset, the others had limb-onset. Dementia was not observed among our C9orf72 mutation carriers.
The heterozygous SOD1-p.Asn87Ser mutation was detected in a sporadic male patient with limb-onset (upper extremity) disease. The age of symptom onset was 48. The patient had man-in-barrel syndrome. Healthy sibs (five sisters and two brothers) and the healthy mother did not carry this variant. The father who died at a car accident at the age of 50 could not be tested; however, he is most probably the transmitter of the variant.
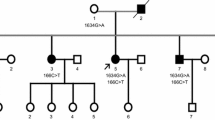
Causative variants are identified in all four fALS cases analyzed by whole exome sequencing. In a family with six affected members (father, four sisters, and nephew), a novel VCP mutation (c.572G > C p.Arg191Pro, NM_007126) was described, which exhibits dominant inheritance characteristics and is accompanied by dementia symptoms (Fig. 1).

Pedigree of familial ALS with VCP mutation (c.572G > C p.Arg191Pro, NM_007126). The index patient is shown by the arrow. Black ALS patient, white unaffected
A heterozygous mutation in the SH3TC2 gene (c.1568 T > C p.Met523Thr, NM_0024577.4) was present in a fALS case. The patient presented with lower limb-onset disease at 46 years of age. An evaluation of the proband’s 52-year-old brother and his 50-year-old sister showed a similar pattern of ALS, with the same heterozygous mutation in the SH3TC2 gene.
A novel homozygous SPG11 frameshift mutation (c.1966_1967delAA, p.Lys656Valfs*11, NM_025137) was detected in two siblings (one male and one female) with juvenile-onset.
Finally, a rare homozygous mutation in TBK1 (c.1928_1930delAAG; p.Glu643del, NM_013254.4) was identified in a fALS case with FTD. The mean age at symptom (weakness of upper extremity) onset was 56. Table 2 summarizes the clinical and genetic data of patients whose diagnosis was confirmed by genetic analysis. Pathogenic variants were not detected in the 25 sALS patients subjected to WGS in the following genes: ALS2, ANG, C19ORF12, CCNF, CHCHD10, CHMP2B, CHRNA3, CHRNA4, CHRNB4, CREST, DAO, DCTN1, ELP3, ERBB4, EWSR1, FIG4, FUS, hnRNPA1, hnRNPA2B1, MATR3, NEFH, NEK1, OPTN, PFN1, PNPLA6, PON1, PON3, PRPH, SETX, SIGMAR1, SOD1, SPG11, SQSTM1, TAF15, TARDBP, TBK1, TUBA4A, UBQLN2, and VAPB.
Discussion
The physiopathological basis of ALS is selective degeneration of motor neurons with a progressive course. The diagnosis of the disease is very often not straightforward and several other diseases have to be ruled out before a firm diagnosis can be ascertained. Thus, the short life span after the establishment of a diagnosis, the absence of a specific treatment for the disease, and the fast clinical progression render molecular genetic investigation of the disease an important issue. Although a description of the differential diagnosis of ALS is clinically not difficult, a variety of underlying pathophysiological processes lead to the premise that ALS pathophysiology is complex with numerous assumptions made so far [30].
The mean age of onset of ALS varies from 50 to 65 years, commonly ranging between 47 and 52 years for fALS [31, 32]. In our study, the mean age at onset for fALS was 49.9 (range: 17–77) years. A study reported that after 80 years of age, the incidence of ALS rapidly decreases, yet, we report two (3.6%) sALS patients whose ages of onset were 82 and 83 [32].
In accordance with the fact that male gender is an established risk factor for ALS, higher incidence of the disease was seen in male patients compared to women (58.2%) also in this cohort [33, 34]. Worldwide, only 5–10% of ALS cases have a family history and are considered as fALS [8]. In our study, fALS cases were higher in incidence compared to the literature (18.2%, 10/55), which may be due to the high rates of consanguineous marriages in Turkey.
Clinical findings of ALS are seen as various combinations of symptoms due to upper and/or lower neuronal damage at the bulbar region and/or limbs. Progressive muscle weakness and atrophy, speech difficulty, difficulty in swallowing and breathing, and fasciculations are signs of lower motor neuron involvement, and spasticity and hyperreflexity are the primary symptoms of upper motor neuron damage. In the later stages, secondary symptoms such as pain, posture disorders, and loss of mobility that all lower the quality of life of patients may be added [19, 35]. The disease onset site observed in the present cohort, as 25.5% bulbar-onset and 72.7% limb-onset, reflects previous studies [36, 37]. Bulbar-onset disease was not seen among our fALS patients. Muscle atrophy was present in all subjects and the distribution of the atrophy was wide in four patients. Progressive weakness and atrophy of upper limbs and absence of bulbar signs are characteristics of the motor neuron man-in-barrel syndrome [38]. A few cases in whom there was respiratory insufficiency, truncal weakness or dementia, might suggest this is random [37]. A patient with bulbar-onset had weakness of neck flexor muscles as a rare symptom of ALS. Bulbar-onset fALS is rare [39]. In our study, all bulbar-onset patients had sALS.
Our study revealed causative variants in eleven patients (20%): The genes carrying these variants were C9orf72 (10.9%), SOD1 (1.8%), VCP (1.8%), SPG11 (1.8%), SH3TC2 (1.8%), and TBK1 (1.8%). Since our cohort covers only the city of Sakarya, our numbers are too small to calculate reliable frequencies; however,, C9orf72 expansion is by far the most abundant ALS cause also in the current Turkish cohort, supporting the findings of a larger epidemiological study on ALS in Turkey with a genetic profile ranking of C9orf72 (18.3%), SOD1 (12.2%), FUS (5%), TARDBP (3.7%), and UBQLN2 (2.4%) in fALS [35, 40].
The north to south decrease in the frequency of C9orf72 expansion across Europe [27] explains the relatively low percentage of C9orf72 in our cohort and in Turkey at large, although it is the leading gene in the country. In our study, only one patient with a C9orf72 expansion had bulbar-onset. The difference of our overall results as compared to the epidemiological data in Turkey may be explained by several factors, including an intense influx of internal migration to Sakarya region, the presence of multi-ethnicity in this relatively small area, a rapid increase of annual resident population, and the relative rarity of consanguineous marriages.
SOD1-based ALS, which represents 20% of fALS and 5% of sALS, is observed in only one case in our cohort [26]. The heterozygous SOD1-p.Asn87Ser variation detected in our sALS patient with an age of symptom onset of 48 was previously reported in another patient with an earlier age of onset of 37 [41]. The two most common SOD1 variants causing ALS in Turkey are reported to be the common Balkan variant, SOD1-p.Leu145Phe, and the homozygous-p.Asp91Ala variant [35]. The absence of these two variants in our cohort is not surprising, since Sakarya is not a region that commonly receives migration from the Balkans. Furthermore, consanguinity in the cohort under study was rare. This observation might partly explain the decreased percentage of SOD1-based ALS in our study. The low number of common fALS mutations is the result of low fALS cases in this study.
In this study, we were able to identify causative variants in all four cases that were analyzed by WES. Two of the cases solved had strong family histories and the other two were offspring to consanguineous parents. The heterozygous VCP variant (p.Arg191Pro) identified in a female fALS patient, also detected in additional five affected family members, caused hallucinations and dementia in addition to ALS symptoms. Mutations in SPG11 are most common cause of hereditary spastic paraplegia [42]. But these mutations can cause rare forms of juvenile-onset ALS as well [43]. In our study, the novel homozygous SPG11 frameshift mutation (c.1966_1967delAA, p.Lys656Valfs*11) was detected in two siblings (one male and one female) with juvenile-onset. Mutations in TBK1 are cause of impaired autophagy and contribute to the accumulation of protein aggregates and ALS pathology [44]. A study reported that TBK1 mutations occurred more frequently in patients with FTD-ALS comorbidity (10.8%) than in patients with ALS alone (0.5%) [45,46,47]. In accordance with this observation, our fALS case with the homozygous TBK1 (p.Glu643del) variant showed FTD symptoms accompanying ALS. The patient’s father and two paternal uncles were reported to have pure FTD and his two sisters died with ALS-FTD. Finally, the heterozygous p.Met523Thr variant in the SH3TC2 gene was detected in a female ALS patient and two affected siblings. In this extended family in which several affected members are described, segregation analysis is ongoing and it cannot be claimed that the above variant in the SH3TC2 gene is the only gene variant responsible of the phenotype. Autosomal dominant mutations in the SH3TC2 gene have been previously associated with mononeuropathy of the median nerve [48].
Genetic variants identified in this small cohort once more point to the genetic heterogeneity well-established in ALS. Further research is pending in cases in whom the underlying genetic defect could not be unraveled yet.
Riluzole and Edaravone are the only two drugs approved by the FDA for ALS, a disease with limited treatment options and no effective therapies [49]. Thus, improving the quality of life of patients with palliative therapies is of utmost importance. In this study, we started riluzole and proper palliative therapies in patients with established diagnoses of ALS. Two patients had also a diaphragm pacing system. During the follow-up period, twenty-three patients died.
Epidemiological studies on ALS may help to understand the effects of environmental factors on disease development and the status of genetic and geographical factors in ALS etiology. Thus, we hope that the clinical and genetic findings presented here contribute to the literature.
Data availability
Data are avaliable.
References
Deenen J, Horlings C, Verschuuren J, Verbeek ALM, van Engelen BGM (2015) The epidemiology of neuromuscular disorders: a comprehensive overview of the literature. J Neuromuscul Dis 2:73–85
Longinetti E, Regodón Wallin A, Samuelsson K, Press R, Zachau A, Ronnevi LO et al (2018) The Swedish motor neuron disease quality registry. Amyotroph Lateral Scler Frontotemporal Degener 19(7–8):528–537. https://doi.org/10.1080/21678421.2018.1497065
Leighton DJ, Newton J, Stephenson LJ, Colville S, Davenport R, Gorrie G et al (2019) Changing epidemiology of motor neurone disease in Scotland. J Neurol 266(4):817–825. https://doi.org/10.1007/s00415-019-09190-7
Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J et al (2018) Prevalence of amyotrophic lateral sclerosis: United States. MMWR Morb Mortal Wkly Rep 67(46):1285–1289. https://doi.org/10.15585/mmwr.mm6746a1
Turgut N, Saraçoğlu GV, Kat S, Balci K, Güldiken B, Birgili O et al (2019) An epidemiologic investigation of amyotrophic lateral sclerosis in Thrace, Turkey, 2006–2010. Amyotroph Lateral Scler Frontotemporal Degener 20(1–2):100–106. https://doi.org/10.1080/21678421.2018.1525403
Chiò A, Logroscino G, Traynor B, Collins J, Simeone JC, Goldstein LA et al (2013) Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41(2):118–140. https://doi.org/10.1159/000351153
Ryan M, Heverin M, McLaughlin RL, Hardiman O (2019) Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol 76(11):1367–1374. https://doi.org/10.1001/jamaneurol.2019.2044
Abhinav K, Stanton B, Johnston C, Hardstaff J, Orrell RW, Clarke J et al (2007) Amyotrophic lateral sclerosis in South-East England: a population-based study. The South-East England register for amyotrophic lateral sclerosis (SEALS Registry). Neuroepidemiology 29(1–2):44–48. https://doi.org/10.1159/000108917
Chiò A, Calvo A, Moglia C, Mazzini L, Mora G, PARALS study group (2011) Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry 82(7):740–746. https://doi.org/10.1136/jnnp.2010.235952
Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377(2):162–172. https://doi.org/10.1056/NEJMra1603471
Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O (1999) Incidence and prevalence of ALS in Ireland, 1995–1997: a population-based study. Neurology 52(3):504–509. https://doi.org/10.1212/wnl.52.3.504
Beghi E, Millul A, Micheli A, Vitelli E, Logroscino G, SLALOM Group (2007) Incidence of ALS in Lombardy, Italy. Neurology 68(2):141–145. https://doi.org/10.1212/01.wnl.0000250339.14392.bb
Vázquez MC, Ketzoián C, Legnani C, Rega I, Sánchez N, Perna A et al (2008) Incidence and prevalence of amyotrophic lateral sclerosis in Uruguay: a population-based study. Neuroepidemiology 30(2):105–111. https://doi.org/10.1159/000120023
Huisman MH, de Jong SW, van Doormaal PT, Weinreich SS, Schelhaas HJ, van der Kooi AJ et al (2011) Population based epidemiology of amyotrophic lateral sclerosis using capture-recapture methodology. J Neurol Neurosurg Psychiatry 82(10):1165–1170. https://doi.org/10.1136/jnnp.2011.244939
Fang F, Valdimarsdottir U, Bellocco R, Ronnevi LO, Sparén L, Fall K et al (2009) Amyotrophic lateral sclerosis in Sweden, 1991–2005. Arch Neurol 66(4):515–519. https://doi.org/10.1001/archneurol.2009.13
Uenal H, Rosenbohm A, Kufeldt J, Weydt P, Goder K, Ludolph A et al (2014) Incidence and geographical variation of amyotrophic lateral sclerosis (ALS) in Southern Germany completeness of the ALS registry Swabia. PLoS ONE 9(4):e93932. https://doi.org/10.1371/journal.pope.0093932
Okamoto K, Hirai S, Yamazaki T, Sun X, Nakazato Y (1991) New ubiquitin-positive intraneuronal inclusions in the extra-motor cortices in patients with amyotrophic lateral sclerosis. Neurosc Lett 129:233–236. https://doi.org/10.1016/0304-3940(91)90469-a
O’Toole O, Traynor BJ, Brennan P, Sheehan C, Frost E, Corr B et al (2008) Epidemiology and clinical features of amyotrophic lateral sclerosis in Ireland between 1995 and 2004. J Neurol Neurosurg Psychiatry 79:30–32. https://doi.org/10.1136/jnnp.2007.117788
Handy CR, Krudy C, Boulis N, Federici T (2011) Pain in amyotrophic lateral sclerosis: a neglected aspect of disease. Neurol Res Int 2011:403808. https://doi.org/10.1155/2011/403808
de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J et al (2008) Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 119(3):497–503. https://doi.org/10.1016/j.clinph.2007.09.143
Ghasemi M, Brown RH Jr (2018) Genetics of amyotrophic lateral sclerosis. Cold Spring Harbour Perspect Med 27:a024125–a024125. https://doi.org/10.1101/cshperspect.a024125
Ajroud-Driss S, Siddique T (1852) 2015) Sporadic and hereditary amyotrophic lateral sclerosis (ALS. Biochim Biophys Acta 4:679–684. https://doi.org/10.1016/j.bbais.2014.08.010
Mulder DW, Kurland LT, Offord KP, Beard CM (1986) Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology 36:511–517. https://doi.org/10.1212/wnl.36.4.511
Renton AE, Chió A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17(1):17–23. https://doi.org/10.1038/nn.3584
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211. https://doi.org/10.1126/science.1165942
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O et al (2011) Amyotrophic lateral sclerosis. Lancet 377:942–955. https://doi.org/10.1016/S0140-6736(10)61156-7
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11:323–330. https://doi.org/10.1016/S1474-4422(12)70043-1
Hardiman O, van den Berg LH, Kiernan MC (2011) Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 7:639–649. https://doi.org/10.1038/nrneurol.2011.153
Fogel BL, Satya-Murti S, Cohen BH (2016) Clinical exome sequencing in neurologic disease. Neurol Clin Pract 6(2):164–176. https://doi.org/10.1212/CPJ.0000000000000239
Gordon PH (2011) Amyotrophic lateral sclerosis: pathophysiology, diagnosis and management. CNS Drugs 25(1):1–15. https://doi.org/10.2165/11586000-000000000-00000
Zarei S, Carr K, Reiley L, Diaz K, Orleiquis G, Altamirano PF et al (2015) A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 6:171. https://doi.org/10.4103/2152-7806.169561
Cronin S, Hardiman O, Traynor BJ (2007) Ethnic variation in the incidence of ALS: a systematic review. Neurology 68(13):1002–1007. https://doi.org/10.1212/01.wnl.0000258551.96893.6f
Logroscino G, Traynor BJ, Hardiman O, Chió A, Mitchell D, Swinger RJ et al (2010) Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry 81(4):385–390. https://doi.org/10.1136/jnnp.2009.183525
Chió A, Moglia C, Canosa A, Umberto M, Dovidio F, Vasta R et al (2020) ALS phenotype is inflenced by age, sex, and genetics. A population-based study. Neurology 94(8):e802–e810. https://doi.org/10.1212/WNL.0000000000008869
Özoguz A, Uyan Ö, Birdal G, Iskender C, Kartal E, Lahut S et al (2015) The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol Aging 36:1764.e9–1764. https://doi.org/10.1016/j.neurobiolaging.2014.12.032
Ralli M, Lambiase A, Artico M, de Vincentiis MGA (2019) Amyotrophic lateral sclerosis: autoimmune pathogenic mechanism, clinical features, and therapeutic perspectives. Isr Med Assoc J 21(7):438–443
Turner MR, Barnwell J, Al-Chalabi A, Eisen A (2012) Young-onset amyotrophic lateral sclerosis: historical and other observations. Brain 35:2883–2891. https://doi.org/10.1016/j.ncl.2015.07.003
Jawdat O, Statland JM, Barohn RJ, Katz JS, Dimachkie MM (2015) Amyotrophic lateral sclerosis regional variants (Brachial Amyotrophic Diplegia, Leg Amyotrophic Diplegia, and Isolated Bulbar Amyotrophic Lateral Sclerosis). Neurol Clin 34:775–785
De Marchi F, Corrado L, Bersano E, Sarnelli MF, Solara V, D’Alfonso S et al (2018) Ptosis and bulbar onset: an unusual phenotype of familial ALS? Neurol Sci 39(2):377–378. https://doi.org/10.1007/s017-3186-0
Millecamps S, Corcia P, Cazeneuve C, Boillee S, Seilhean D, Danel-Brunaud V et al (2012) Mutations in UBQLN2 are rare in French amyotrophic lateral sclerosis. Neurobiol Aging 33(839):e1–3. https://doi.org/10.1016/j.neurobiolaging.2011.11.010
Khani M, Alavi A, Nafissi S, Elahi E (2015) Observation of c.260A > G mutation in superoxide dismutase 1 that causes p.Asn86Ser in Iranian amyotrophic lateral sclerosis patient and absence of genotype/phenotype correlation. Iran J Neurol 14(3):152–157
Bouslam N, Bouhouche A, Benomar A, Hanein S, Klebe S, Azzedine H et al (2007) A novel locus for autosomal recessive spastic ataxia on chromosome 17p. Hum Genet 121(3–4):413–420. https://doi.org/10.1007/s00439-007-0328-0
Daoud H, Zhou S, Noreau A, Sabbagh M, Belzil V, Dionne-Laporte A et al (2012) Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging 33(4):839. https://doi.org/10.1016/j.neurobiolaging.2011.11.012
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS et al (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347(6229):1436–1441. https://doi.org/10.1126/science.aaa3650
Le Ber I, De Septenville A, Millecamps S, Camuzat A, Caroppo P, Couratier P et al (2015) TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging 36(11):3116.e3115–3118. https://doi.org/10.1016/j.neurobiolaging.2015.08.009
Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Philtjens S, Heeman B et al (2015) Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology 85(24):2116–2125. https://doi.org/10.1212/WNL.0000000000002220
Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R et al (2015) Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 130(1):77–92. https://doi.org/10.1007/s00401-015-1436-x
Arnaud E, Zenker J, de Preux Charles AS, Stendel C, Roos A, Médard JJ et al (2009) SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc Natl Acad Sci USA 106:17528–17533. https://doi.org/10.1073/pnas.0905523106
Bensimon G, Lacomblez L, Delumeau JC, Bejuit R, Truffinet P, Meininger V et al (2002) A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol 249:609–615. https://doi.org/10.1007/s004150200071
Acknowledgement
NDAL is grateful to Suna and İnan Kıraç Foundation for the generous support of the study and for the inspiring research environment created at Koç University-KUTTAM. We thank Elif Bayraktar for her assistance in drawing the pedigree.
Funding
The genetic analyses of this study were financed by Suna and İnan Kıraç Foundation at Koç University-KUTTAM.
Author information
Authors and Affiliations
Contributions
ZÖA, DK, CT, FA, ANB provided author’s contributions concepts. DK, ZÖA, CT, FA, ANB involved in design. ZÖA, DK, BDG, CT, FA, ANB contributed materials. ZÖA, DK, BDG, CT, FA, ANB collected the data. ZÖA, CT, DK, ANB performed literature search. ZÖA, CT, DK, ANB wrote the manuscript. ANB, ZÖA, CT, DK involved in critical reviews.
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest was declared by the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kotan, D., Özözen Ayas, Z., Tunca, C. et al. Phenotypic and genotypic features of patients diagnosed with ALS in the city of Sakarya, Turkey. Acta Neurol Belg 120, 1411–1418 (2020). https://doi.org/10.1007/s13760-020-01441-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-020-01441-z