
Abstract
Migonemyia migonei (Franҫa, 1920) (Diptera: Psychodidae) belongs to the subfamily Phlebotominae, of epidemiological importance due to its role as a vector in leishmaniasis transmission cycles and its broad geographic distribution in South America. Few morphometric and genetic studies have demonstrated the existence of variability among geographically distant populations in Brazil. The aim of the study was to estimate the genetic distance within the morphospecies Mg. migonei through the analysis of cytochrome C oxidase subunit I (COI) sequences of specimens captured in Argentina and those available in online databases. The COI sequences from specimens collected in different localities of Argentina and sequences available in online databases were utilized. Genetic distances were analyzed and a median-joining haplotype network was constructed. Finally, phylogenetic reconstruction was performed according to Bayesian inference. The analyses led to the identification of at least two haplogroups: haplogroup I with sequences of specimens from Colombia, Brazil and Argentina, and haplogroup II with sequences of specimens from Argentina. Interestingly, specimens from Argentina whose haplotypes corresponded to both haplogroups, were collected in sympatry. The results suggest that Mg. migonei could be a species complex with at least two distinct members. This hypothesis could explain the known characteristics of adaptability and vector permissiveness of the species, as the putative cryptic species of the complex could differ in traits of epidemiological importance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Migonemyia migonei (Franҫa, 1920) is a Phlebotominae species (Diptera: Psychodidae) that is widely distributed in South America from Colombia to Argentina. It belongs to the subgenus Migonemyia (Migonemyia), which includes the species Mg. rabelloi (Galati & Gomes, 1992) and Mg. vaniae Galati, Fonseca & Marassá, 2007, both with a geographic distribution restricted to São Paulo, Brazil (Galati 2018). The species Mg. migonei is described as exhibiting zoophilic behavior, but it also exhibits the ability to adapt to modified environments, thus presenting relative anthropophilic behavior (Salomón et al. 2010; Aguiar et al. 2014). It is considered to be of medical importance because of its role as a vector in leishmaniases transmission cycles. Leishmaniases are a group of neglected tropical diseases, endemic in 18 countries in the American continent. More than 1 million cases of tegumentary leishmaniasis (TL) and 67,000 cases of visceral leishmaniasis (VL) have been reported in the last 20 years (Organización Panamericana de la Salud 2021). Its epidemiology comprises a variety of transmission scenarios that differ according to the species involved in both the Phlebotominae vector and the Leishmania Ross, 1903 parasite (Organización Panamericana de la Salud 2019). In order to be confirmed as a vector, a Phlebotominae species must meet the criteria defined by Killick-Kendrick (1990) which include, for example, ecological aspects such as anthropophilic behavior, feeding preference for the genus Leishmania reservoirs, and a geographical distribution consistent with the area of disease occurrence. In turn, the identification and classification of each transmission scenarios is necessary to know the magnitude and risk of the disease occurrence, allowing the recognition of geographical areas with and without transmission, and the planning and orientation of surveillance and control actions (Organización Panamericana de la Salud 2019). Although mainly associated with TL transmission cycles (Aguiar and Vieira 2018; Rangel et al. 2018), Mg. migonei has also been indicated as a putative vector of Leishmania infantum Nicolle, 1908 (the etiological agent of VL) in an enzootic cycle with accidental human transmission in Santiago del Estero, Argentina (Salomón et al. 2010), and in Pernambuco, Brazil (Carvalho et al. 2007), both situations without the occurrence of the primary vector Lutzomyia longipalpis (Lutz & Neiva, 1912) (Diptera: Psychodidae). In Argentina, it is the second most abundant species, covering 12 provinces, where it has been collected with high relative density in association with human leishmaniasis cases (Moya et al. 2021).
Despite its epidemiological importance and broad geographic distribution, there are few studies on this species. Based on morphology (Galati et al. 2007), molecular markers (Rodrigues et al. 2018), or a combination of these approaches (Costa et al. 2018), all studies detected some degree of genetic distance between allopatric populations: Galati et al. (2007) analyzed five populations from different regions of Brazil and Peru by morphometry, finding significant differences for the northeastern Brazilian population; Costa et al. (2018) analyzed the genetics and morphology of three populations (two from the northeast and one from the southeast) and concluded the existence of at least two distinct lineages; Rodrigues et al. (2018) also detected cryptic diversity in geographically distant populations, suggesting that the genetic distance found may be due to microevolutionary processes driven by isolation by distance. Given this background, we aimed to estimate the genetic distance in the morphospecies Mg. migonei through the analysis of cytochrome C oxidase subunit I (COI) sequences of specimens captured in Argentina, combined with sequences publicly available in online databases.
Materials and methods
Study area and Phlebotominae sampling
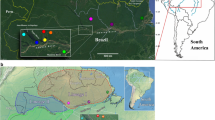
The Phlebotominae were caught with REDILA-BL light traps (Fernández et al. 2015) operating overnight from 18:00 to 8:00 h in the following localities: Yuto (23º38’S, 64º28’W, Jujuy province), Monteros (27º09’S, 65º30’W, Tucumán province), Laishi (26º14’S, 58º38’W, Formosa province) and Fracrán (26º44’S, 54º17’W), Lanusse (25º58’S, 54º15’W), Paraje Mbiguá (27º09’S, 53º57’W), Puerto Iguazú (25º36’S, 54º34’W) and San Pedro (26º38’S, 54º08’ W, Misiones province) (Fig. 1). The captures were carried out as part of the Leishmaniasis Research Network in Argentina research projects, which included the detection of parasites, so all but one of the specimens were females. The thorax and abdomen of the Phlebotominae were dissected and preserved in 90% alcohol for subsequent DNA extraction. The head and the last segments of the abdomen were rinsed and mounted on slides for microscopic observation of taxonomic characters. The species determination was performed according to Galati's key (Galati 2018), and the generic abbreviations follow the proposal of Galati et al. (2017).

Collection sites of Mg. migonei specimens and the COI haplotypes identified. The symbols represent localities covered by different authors: Costa et al. (circle), Pinto et al. (star), Romero-Ricardo et al. (triangle), Laurito et al. (square), and this study (rhombus). More information on number of sequences available is presented in Table 1 and supplementary data
DNA extraction, COI amplification and sequencing
DNA extractions were performed using commercial kits DNA Puriprep-S INBIO HW (Tandil, B.A., Argentina) and Transgen Biotech (Beijing, China) from the preserved tissue of the specimens identified as Mg. migonei. The extraction yield was evaluated by quantification with Qubit™ (Waltham, MA, USA) fluorometer and PCR amplification of the cacophony gene, which is constitutive of the Diptera genome. Amplification of COI was performed according to the protocol described by Hebert et al. (2003), using the primers LCO1490-HCO2198. The PCR was optimized for a final volume of 50 μl (Taq polymerase buffer 1 X, Magnesium Chloride 2.5 M, DMSO 2.5%, dNTPs 0.2 mM, primers 0.5 μM, Taq polymerase 1.4 U) and 5 μl of genomic DNA. Cycling conditions were set to an initial denaturation at 94 ºC for 4 min, 35 cycles of: 30 s at 94 ºC, 30 s at 56 ºC and 1 min at 72 ºC, and a final extension of 10 min at 72 ºC. All amplifications were performed in an Applied Biosystems 2720 thermal cycler (Foster City, CA, USA). The products obtained were separated on a 2% agarose gel stained with SYBR® Safe DNA (Invitrogen, Carlsbad, CA, USA) and visualized with 470 nm blue light. The size of the fragments was compared with a molecular weight marker in the range of 100 to 1000 base pairs (bp), with increments of 100 by 100 bp. From each amplified product, sequencing of both complementary DNA strands was performed on an ABI 3730XLs kit (Macrogen, Seoul, South Korea) or at the Centre of Biodiversity Genomics, University of Guelph, Canada.
Phylogenetic analyses
The DNA sequences were manually aligned using the software MEGA v.10 (Kumar et al. 2018). In addition, sequences available in GenBank and BOLD (Ratnasingham and Hebert 2007) for Mg. migonei were downloaded. A multiple alignment was generated with the CLUSTAL W algorithm (Thompson et al. 1994), and the estimation of pairwise and mean Kimura 2-parameter genetic distances (K2p) both intraspecific and between haplogroups (see results) was performed in MEGA v.10 with 1000 replicates. The haplotypes were estimated using DNaSP v.5.10 (Rozas et al. 2017), and named according to the numbering of the already published sequences. The relationship among haplotypes was evaluated by constructing a minimum haplotype network using the median-joining method implemented in Network v.4.6 (Bandelt et al. 1999), assuming epsilon to be 0, and transition/transversion were given weights of 1 and 2, respectively. For phylogenetic inference, the best-fit model of evolution was estimated using the Bayesian Criterion (BIC), implemented in JModeltest v.2 (Darriba et al. 2012) with the following settings: Phyml version = 2.4.4; candidate models = 24; number of substitution schemes = 3: (1) Including models with equal/unequal base frequencies (+F), (2) Including models with/without a proportion of invariable sites (+I), (3) Including models with/without rate variation among sites (+G) (nCat = 4); optimized free parameters (K) = substitution parameters + 115 branch lengths; base tree for likelihood calculations = BIONJ tree. The Bayesian inference was analyzed using MrBayes: Bayesian Inference of Phylogeny v3.2 (Ronquist et al. 2012), four Monte Carlo Chains were run for 10,000,000 generations (sampled every 1,000 generations) to allow adequate time for convergence (0.006262). The posterior information of topologies and the median branch lengths from the trees sampled were then visualized with FigTree v.1.4.2 (Rambaut 2012). Sequences of Lu. longipalpis (BOLD: PHLAR069-18) and Phlebotomus papatasi Scopoli, 1786 (Diptera: Psychodidae) (GenBank accession: KY848828.1) were used as outgroups.
Results
The dataset consisted of 74 COI sequences of Mg. migonei: 12 obtained from specimens captured in different provinces of Argentina and sequences downloaded from the databases of specimens from Brazil (n = 58) (Pinto et al. 2015; Costa et al. 2018), Colombia (n = 2) (Romero-Ricardo et al. 2016), and two sequences from Argentina (Laurito et al. 2019) (Table 1 and Online Resource 1). No insertions or deletions were observed in the final alignment of 658 bp with 72 variable sites (10.9%). The mean intraspecific K2p genetic distance of Argentina specimens was 1.47% (maximum = 3.69%), reaching 2.03% (max = 5.1%) when considering the entire dataset (global). The genetic distance between haplogroups (see below) was 3.89%. Six new haplotypes were identified (H51-H56) in this study, with GenBank accessions: OR004818-OR004823. A total of 57 haplotypes were identified, the median-joining network showed two haplogroups separated by at least 18 mutational steps: in haplogroup I, there were 49 haplotypes from Brazil, Colombia (H50), and Argentina (H52, in Misiones province). On the other hand, haplogroup II grouped only haplotypes from Argentina (Córdoba, Formosa, Tucumán, Misiones and Jujuy provinces (H51, H53-H57) (Fig. 2a). Haplotype 52 was found at least eight mutational steps away from the Brazilian haplotypes (Pernambuco). Notably, H52 and H53 were found at the same locality in Puerto Iguazú, Misiones, Argentina.

a. Median-joining haplotype network of Mg. migonei based on 658 nucleotides of the COI gene. The circle size corresponds to the frequency of each haplotype in the total number of individuals sampled. Missing haplotypes are shown as grey circles. Each line connecting haplotypes represents one mutational step, whereas numbers along the lines are the total number of mutational steps among haplotypes. b. Bayesian Inference (BI) topology tree from 658 nucleotides of the COI gene, inferred under the HKY + I + G model. Number on each branch (above branch) represent posterior probabilities (PP) obtained in the BI. The scale bar represents the expected number of nucleotide substitutions per site. The asterisks indicate haplotypes from Argentina. The circles at the branch terminals represent the number of individuals of each haplotype. Color references: Argentina: Misiones (light green), Formosa (yellow), Tucumán (orange), Jujuy (red) and Córdoba (brown); Brazil: Espirito Santo (pink), Ceará (turquoise), Pernambuco (blue), and Río de Janeiro (purple); Colombia: Sucre (dark green)
For the Bayesian inference the most appropriate model was HKY + I + G (-InL = 2250.5022, BIC = 5286.1983, Delta BIC = 0, p-inv = 0.6640, Gamma = 0.7560). Two monophyletic clades coincident with the described haplogroups were observed with a posterior probability (PP) = 1.0. The specimens of haplogroup I were grouped with a value of 0.66 PP, along with haplotypes from Colombia, Brazil, and Argentina; while the haplogroup II grouped with a 0.95 PP value, consisting of haplotypes only from Argentina (Fig. 2b).
Discussion
The present study assessed the genetic distance within the species Mg. migonei by analyzing sequences of a fragment of the COI gene from specimens captured in Argentina and those available in online databases. The intraspecific distance obtained from specimens from different provinces of Northern Argentina (mean = 1.47%, max = 3.69%) was higher than that reported by Pinto et al. (2015) with Mg. migonei specimens from different localities of Espírito Santo, Brazil (mean = 0.47%, max = 1.54%), probably due to the difference in the geographic areas covered. It was also higher than that reported by Rodrigues et al. (2018) (1.3%) when comparing those Espírito Santo sequences with their Maranhão haplotypes (northeastern Brazil), probably due to the small number of sequences from Maranhão. In contrast, with a larger number of individuals analyzed from the same Brazilian regions, Costa et al. (2018) described two monophyletic lineages with 3.15% of genetic distance between them, a maximum rate similar to our results. These results are expected since the intraspecific variation estimated and the probability of non-monophyly increases with the larger geographic area covered due to evolutionary processes. It should be noted that the estimation of the sample size needed to capture 95% of intraspecific variation is up to 70 individuals, even if samples are collected maximizing geographic coverage (Bergsten et al. 2012). This situation was attainable since the molecular marker COI is being used in barcoding studies, which implies a high availability of sequences in databases for researchers (Rodrigues and Galati 2023).
For the global analyses conducted in this study, we added new haplotypes to the COI intraspecific variation reported for Mg. migonei while also extended the geographic area covered in previous studies. The maximum pairwise distance obtained was between haplotypes H25 and H53 from Rio de Janeiro (southeastern Brazil) and Misiones (northeastern Argentina) with a genetic distance of 5.1%, although there are sequences that belong to specimens from more geographically distant captured sites. The haplotype 52 is the only haplotype from Argentina that is part of the same haplogroup as all the haplotypes from Brazil and Colombia (haplogroup I) with at least 29 mutational steps of separation between the closest Argentine haplotype (haplogroup II). Interestingly, the H52 was detected in individuals captured in three different localities of Misiones province and in one of them (Puerto Iguazú), the specimen was collected in the same sample site as a specimen with H53, with which it differs by 3.36% in genetic distance and at least by 31 mutational steps. Therefore, the genetic distance between haplotypes captured in sympatry in Misiones province was higher than that reported by Costa et al. (2018) between their geographic linages (3.15% and at least 12 mutational steps). In our results, those Costa lineages were recovered as part of haplogroup I containing also the haplotypes from Colombia, H52 from Argentina and most of the haplotypes from Espirito Santo (Brazil) presenting internal polytomies. With respect to other Phlebotominae species, the genetic distance between haplogroups I and II (3.9% and 29 mutational steps) is higher than that found between the close species Nyssomyia intermedia (Lutz & Neiva, 1912)—Ny. neivai (Pinto, 1926) (3.5%, 11 mutational steps), and Ny. intermedia—Ny. whitmani (Antunes & Coutinho, 1939) (3.4%, 22 mutational steps) as well as between Lutzomyia cruzi (Mangabeira, 1938)—Lu. alencari Martins, Souza & Falҫao, 1962 (3%, 16 mutational steps) (Moya et al. 2020).
About the locality where we detected the haplogroups of Mg. migonei in sympatry, a study of genetic diversity according to gene fragments of the nicotinamide dinucleotide dehydrogenase subunit 4 and the 3’ region of cytochrome B of specimens of the Lu. longipalpis complex found that they belonged to one of the three haplogroups described for this species in Argentina (Pech-May et al. 2018). Another study analyzed COI sequences of Lu. longipalpis complex and Ny. whitmani from the same locality and also did not detect more than one haplogroup in sympatry for each species (Moya et al. 2020). Therefore, if a hypothesized speciation process occurred due to some characteristic of the area, it seems that these other species were not affected in the same way. At this locality these species differ in abundance and seasonal-spatial distribution, so microevolutionary processes will influence each of them differently (Quintana et al. 2020).
Concerning the morphology, distinguishable structures make it possible to differentiate the morphospecies Mg. migonei from its congeners (Galati et al. 2007). Besides other characters, the number of bristles on the eighth abdominal tergite and the shapes of the head of the spermathecae, allowed us to discard errors in the determination of the specimens for which we were able to recheck the morphology. On the other hand, speciation is not always accompanied by morphological change, differences in copulatory courtship song or sex pheromones as described for other cryptic species cannot be ruled out (Bickford et al. 2007; Sousa-Paula et al. 2021). Still, some subtle morphological differences could be detected by morphometric studies to be carried out. These results will be reinforced if characteristics of different types, such as those proposed in integrative taxonomy approaches, are taken into account.
Several epidemiological studies show interesting differences in ecological characteristics of Mg. migonei. In western Venezuela, it was described as being anthropophilic and adapted to both humid and dry mountain forests with a wide altitudinal distribution (Chaves and Añez 2004), and was indicated as a vector in two epidemiological cycles, involving Leishmania mexicana Biagi, 1953 and Le. guyanensis Floch, 1954 (TL etiological agents) (Torrellas et al. 2018). In northeastern Brazil, Mg. migonei was found in high abundance in remote indigenous villages in a semiarid region associated with transmission cycles of Le. braziliensis Vianna, 1911 (TL agent) (Sales et al. 2019) and also in the Atlantic coast adapted to the indoor environment suspected to be involved in Le. infantum (VL agent) transmission (Silva et al. 2014). In southeast Brazil, this species was captured both in peri-urban areas and in wild habitats exhibiting eclecticism in terms of feeding preference (wild rodents, hamsters and chickens) (Taniguchi et al. 2002). In the same region, in a peri-urban area with canine VL cases, its vector capacity was assessed finding that it has cynophilic behavior and the results reinforce the suspicion that it is a potential vector of Le. infantum (Galvis-Ovallos et al. 2017). In Brazil, Mg. migonei is considered a permissive vector with demonstrated susceptibility to the development of different Leishmania species (Nieves and Pimenta 2002; Silva et al. 2014; Guimarães et al. 2016). The DNA of Le. braziliensis and of Le. infantum were detected in specimens captured in Chaco and Misiones provinces (Argentina) respectively, but without confirmation of vectorial capacity (Moya et al. 2021). In Argentina, Mg. migonei is prevalent in the Dry Chaco region, mainly in peri-urban rural transition habitats with domestic animals where it could act as a hinge between zoonotic cycles and sporadic human VL cases (Salomón et al. 2008, 2010; Quintana et al. 2012). However, in the site where both haplogroups were found sympatrically (Misiones, Argentina), Mg. migonei was not abundant (2.8%, data not shown) and the environment was characterized as urban with presence of chickens, domestic animals and a variety of fruit trees (Quintana et al. 2020). It has been also described as zoophilic and has shown a feeding preference for horses (Salomón et al. 2008). The probable avidity for equines was also described in north and southeast Brazil at rural areas largely deforested with occurrence of TL (Rio de Janeiro (Souza et al. 2001) and Ceará States (Azevedo and Rangel 1991)). Interesting, horses are also suspected to be reservoirs of both Le. braziliensis (Truppel et al. 2014) and Le. infantum (Benassi et al. 2018) and mixed infections have been detected (Soares et al. 2013). Meanwhile, it is also suggested that female Phlebotominae are opportunistic and probably adjust their feeding habits to the available hosts, especially when adapting to anthropic environments (Muniz et al. 2006; Afonso et al. 2012).
Until more evidence is gathered, it should be noted that Mg. migonei was described as plastic in its ability to adapt to modified environments, eclectic in terms of feeding preference, and as permissive in terms of vector competence (Salomón et al. 2010; Guimarães et al. 2016; Aguiar and Vieira 2018; Moya et al. 2021). This, combined with its wide geographic distribution, could lead us to consider it as a generalist species. If so, it would not be the first time that, after further study, a generalist species ends up being a complex of specialists, as verified for Lu. longipalpis, Nyssomyia umbratilis (Ward & Fraiha, 1977) and Psathyromyia shannoni (Dyar, 1929) (Bickford et al. 2007; Pinto et al. 2015). Although the pattern of evolution of a single gene will not necessarily correspond to the history of the species analyzed, our results support the hypothesis of cryptic diversity in Mg. migonei, suggesting that it may be a species complex with at least two distinct haplogroups. While the detection of genetic differences in allopatric populations could be ascribable to local adaptations or genetic drift, its detection in sympatry suggests that some degree of reproductive isolation may exist (Bickford et al. 2007). These putative cryptic species could differ in the following traits with different impact on leishmaniasis transmission: (i) anthropophilia/zoophilia (equinophilia or cynophilia), allowing the identification of potential reservoirs and attractors; (ii) adaptability to wild/anthropic environment, for the definition of areas at risk of transmission, the human population exposed and the possibility of maintenance of parasites circulating in interepidemic zoonotic cycles; (iii) permissive/specific vector competence, for the identification of the etiological agent involved and its geographic area of occurrence or potential expansion. Therefore, for experimental studies on vector competence and insecticide resistance, it would be important to determine the membership of the test specimens to the described haplogroups in order not to make inferences sensu lato that cannot be extrapolated to the different members of the complex, as was the case with Lu. longipalpis. In turn, the definition of these aspects of the vector contributes to the identification of different transmission scenarios, allowing the risk and magnitude of occurrence of leishmaniasis to be known. If the existence of the complex is proven, correctly distinguishing between its members and identifying their respective geographic distribution will enable the direction of efforts towards ensuring that prevention and control measures against leishmaniases are cost-effective and appropriate.
References
Afonso MMS, Chaves SAM, Rangel EF (2012) Evaluation of feeding habits of haematophagous insects, with emphasis on Phlebotominae (Diptera: Psychodidae), vectors of Leishmaniasis. Trends Entomol 8:125–136
Aguiar GM, Vieira VR (2018) Regional distribution and habitats of Brazilian Phlebotomine species. In: Rangel EF, Shaw JJ (eds) Brazilian Sand Flies, 1st edn. Springer, Switzerland, pp 251–298
Aguiar GM, Azevedo ACR, Medeiros WM, Alves JRC, Rendeiro V (2014) Aspects of the ecology of Phlebotomines (Diptera: Psychodidae: Phlebotominae) in an area of cutaneous leishmaniasis occurrence, Municipalitu of Angra dos Reis, Coast of Rio de Janeiro State, Brazil. Rev Inst Med Trop São Paulo 56:143–149. https://doi.org/10.1590/S0036-46652014000200010
Azevedo ACR, Rangel EF (1991) A study of sandfly species (Diptera: Psychodidae: Phlebotominae) in a focus of cutaneous leishmaniasis in the municipality of Baturité, Ceará, Brazil. Mem Inst Oswaldo Cruz 86:405–410
Bandelt H-J, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1146/annurev.es.18.110187.002421
Benassi JC, Benvenga GU, Ferreira HL, Soares RM, Silva DT, Pereira VF, Ruiz VLA, Oliveira TMFS (2018) Molecular and serological detection of Leishmania spp. in horses from an endemic area for canine visceral leishmaniasis in southeastern Brazil. Pesq Vet Bras 38:1058–1063. https://doi.org/10.1590/1678-5150-PVB-5214
Bergsten J, Bilton DT, Fujisawa T, Elliott M, Monaghan MT, Balke M, Hendrich L, Geijer J, Herrmann J, Foster GN, Ribera I, Nilsson AN, Barraclough TG, Vogler AP (2012) The effect of geographical scale of sampling on DNA barcoding. Syst Biol 61:851–869. https://doi.org/10.1093/sysbio/sys037
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, Ingram KK, Das I (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22:148–155. https://doi.org/10.1016/j.tree.2006.11.004
Carvalho MR, Lima BS, Marinho-Júnior JF, Silva FJ, Valenҫa HF, Almeida FA, Silva AL, Brandão-Filho SP (2007) Phlebotomine sandfly species from an American visceral leishmaniasis area in the Northern Rainforest region of Pernambuco State, Brazil. Cad Saude Publica 23:1227–1232. https://doi.org/10.1590/s0102-311x2007000500024
Chaves LF, Añez N (2004) Species co-occurrence and feeding behavior in sand fly transmission of American cutaneous leishmaniasis in western Venezuela. Acta Trop 92:219–224. https://doi.org/10.1016/j.actatropica.2004.08.001
Costa PL, Brazil RP, Fuzari AA, Latrofa MS, Annoscia G, Tarallo VD, Capelli G, Otranto D, Brandão-Filho SP, Dantas-Torres F (2018) Morphological and phylogenetic analysis of Lutzomyia migonei from three Brazilian states. Acta Trop 187:144–150. https://doi.org/10.1016/j.actatropica.2018.07.027
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and high-performance computing Europe. PMC Funders Group Nat Methods 9:772. https://doi.org/10.1038/nmeth.2109
Fernández MS, Martínez MF, Pérez AA, Santini MS, Gould IT, Salomón OD (2015) Performance of light-emitting diode traps for collecting sand flies in entomological surveys in Argentina. J Vector Ecol 40:373–378. https://doi.org/10.1111/jvec.12176
Galati EAB (2018) Phlebotominae (Diptera, Psychodidae): classification, morphology and terminology of adults and identification of american taxa. In: Rangel EF, Shaw JJ (eds) Brazilian sand flies, 1st edn. Springer, Switzerland, pp 9–212
Galati EAB, Fonseca MB, Marassá AM (2007) The subgenus Migonemyia Galati 1995 (Diptera, Psychodidae, Phlebotominae), with description of a new species Migonemyia vaniae. Mem Inst Oswaldo Cruz 102:605–615. https://doi.org/10.1590/S0074-02762007005000064
Galati EAB, Galvis-Ovallos F, Lawyer P, Léger N, Depaquit J (2017) An illustrated guide for characters and terminology used in descriptions of Phlebotominae (Diptera, Psychodidae). Parasite 24:1–35. https://doi.org/10.1051/parasite/2017027
Galvis-Ovallos F, da Silva MD, Bispo GBDS, de Oliveira AG, Neto JRG, Malafronte RDS, Galati EAB (2017) Canine visceral leishmaniasis in the metropolitan area of São Paulo: Pintomyia fischeri as potential vector of Leishmania infantum. Parasite 24:2. https://doi.org/10.1051/parasite/2017002
Guimarães VCFV, Pruzinova K, Sadlova J, Volfova V, Myskova J, Brandão-Filho SP, Volf P (2016) Lutzomyia migonei is a permissive vector competent for Leishmania infantum. Parasit Vectors 9:1–6. https://doi.org/10.1186/s13071-016-1444-2
Hebert PDN, Ratnasingham S, deWaard JR (2003) Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond B (Suppl) 270:S96–S99. https://doi.org/10.1098/rsbl.2003.0025
Killick-Kendrick R (1990) Phlebotomine vectors of the leishmaniases: a review. Med Vet Entomol 4:1–24
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Laurito M, Ontivero IM, Almirón WR (2019) Increasing the digital repository of DNA barcoding sequences of sand flies (Psychodidae: Phlebotominae). Mem Inst Oswaldo Cruz 114:1–5. https://doi.org/10.1590/0074-02760190208
Moya SL, Pech-May A, Quintana MG, Manteca-Acosta M, Salomón OD (2020) Phylogenetic relationships of closely-related phlebotomine sand flies (Diptera: Psychodidae) of Nyssomyia genus and Lutzomyia subgenus. Mem Inst Oswaldo Cruz 115:1–10. https://doi.org/10.1590/0074-02760200220
Moya SL, Szelag EA, Manteca-Acosta M, Quintana MG, Salomón OD (2021) Update of the Phlebotominae fauna with new records for Argentina and observations on leishmaniasis transmission scenarios at a regional scale. Neotrop Entomol 51:311–323. https://doi.org/10.1007/s13744-021-00934-7
Muniz LHG, Rossi RM, Neitzke HC, Monteiro WM, Teodoro U (2006) Host feeding preferences of sandflies in rural area, Southern Brazil. Rev Saúde Pública 40:1087–1093
Nieves E, Pimenta PFP (2002) Development of Leishmania (Viannia) braziliensis and Leishmania (Leishmania) amazonensis in the sand fly Lutzomyia migonei (Diptera: Psychodidae). Am J Trop Med Hyg 67:640–647. https://doi.org/10.4269/ajtmh.2002.67.640
Organización Panamericana de la Salud (2019) Manual de procedimientos para la vigilancia y control de las leishmaniasis en las Américas. Washington, D.C. https://doi.org/10.37774/9789275320631. Accessed 24 Feb 2023
Organización Panamericana de la Salud (2021) Leishmaniasis: Informe epidemiológico de las Américas. https://iris.paho.org/handle/10665.2/55368. Accessed 14 March 2023
Pech-May A, Ramsey J, González Ittig R, Giuliani M, Berrozpe P, Quintana MG, Salomón OD (2018) Genetic diversity, phylogeography and molecular clock of the Lutzomyia longipalpis complex (Diptera: Psychodidae). PLoS Negl Trop Dis 12:1–22. https://doi.org/10.1371/journal.pntd.0006614
Pinto IDS, Chagas BDd, Rodrigues AAF, Ferreira AL, Rezende HR, Bruno RV, Falqueto A, Andrade-Filho JD, Galati EAB, Shimabukuro PHF, Brazil RP, Peixoto AA (2015) DNA barcoding of neotropical sand flies species identification and discovery within Brazil. PLoS ONE 10:1–18. https://doi.org/10.1371/journal.pone.0140636
Quintana MG, Fernández MS, Salomón OD (2012) Distribution and abundance of Phlebotominae, vectors of Leishmaniasis, in Argentina: spatial and temporal analysis at different scales. J Trop Med 2012:1–16. https://doi.org/10.1155/2012/652803
Quintana MG, Santini MS, Cavia R, Martínez MF, Liotta DJ, Fernández MS, Pérez AA, Mancini JMD, Moya SL, Giuliani MG, Salomón OD (2020) Multiscale environmental determinants of Leishmania vectors in the urban-rural context. Parasit Vectors 13:1–15. https://doi.org/10.1186/s13071-020-04379-6
Rambaut A (2012) FigTree. (version 1.4.4). Available at: http://tree.bio.ed.ac.uk/software/figtree/. Accessed 10 Apr 2023
Rangel EF, Lainson R, Carvalho BM, Costa SM, Shaw JJ (2018) Sand fly vectors of American cutaneous leishmaniasis in Brazil. In: Rangel EF, Shaw JJ (eds) Brazilian sand flies, 1st edn. Springer, Switzerland, pp 341–380
Ratnasingham S, Hebert PDN (2007) BARCODING, BOLD: the barcode of life data system (www.barcodinglife.org). Mol Ecol Notes 7:355–364. https://doi.org/10.1111/j.1471-8286.2006.01678.x
Rodrigues BL, Carvalho-Costa LF, Pinto IDS, Rebêlo JMM (2018) DNA barcoding reveals hidden diversity of sand flies (Diptera:Psychodidae) at fine and broad spatial scales in Brazilian endemic regions for Leishmaniasis. J Med Entomol 55:893–901. https://doi.org/10.1093/jme/tjy032
Rodrigues BL, Galati EAB (2023) Molecular taxonomy of phlebotomine sand flies (Diptera, Psychodidae) with emphasis on DNA barcoding: a review. Acta Trop 238. https://doi.org/10.1016/j.actatropica.2022.106778
Romero-Ricardo L, Lastre-Meza N, Pérez-Doria A, Bejarano EE (2016) DNA barcoding to identify species of phlebotomine sand fly (Diptera: Psychodidae) in the mixed leishmaniasis focus of the Colombian Caribbean. Acta Trop 159:125–131. https://doi.org/10.1016/j.actatropica.2016.03.017
Ronquist F, Teslenko M, Mark PVD, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) Mrbayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Sales KGDS, De Oliveira Miranda DE, Costa PL, Silva JF, Figueredo LA, Brandão-Filho SP, Dantas-Torres F (2019) Home sweet home: Sand flies find a refuge in remote indigenous villages in north-eastern Brazil, where leishmaniasis is endemic. Parasit Vectors 12:1–12. https://doi.org/10.1186/s13071-019-3383-1
Salomón OD, Rosa JR, Stein M, Quintana MG, Fernández MS, Visintin AM, Spinelli GR, Pascual MMB, Molinari ML, Morán ML, Valdez D, Bruno MR (2008) Phlebotominae (Diptera: Psycodidae) fauna in the Chaco region and Cutaneous Leishmaniasis transmission patterns in Argentina. Mem Inst Oswaldo Cruz 103:578–584. https://doi.org/10.1590/S0074-02762008000600011
Salomón OD, Quintana MG, Bezzi G, Morán ML, Betbeder E, Valdéz DV (2010) Lutzomyia migonei as putative vector of visceral leishmaniasis in La Banda, Argentina. Acta Trop 113:84–87. https://doi.org/10.1016/j.actatropica.2009.08.024
Silva RA, Santos FKM, de Sousa LC, Rangel EF, Bevilaqua CML (2014) Ecology of Lutzomyia longipalpis and Lutzomyia migonei in an endemic area for visceral leishmaniasis. Rev Bras Parasitol Veterinária 23:320–327. https://doi.org/10.1590/s1984-29612014068
Soares IR, Silva SO, Moreira FM, Prado LG, Fantini P, Maranhão RPA, Silva Filho JM, Melo MN, Palhares MS (2013) First evidence of autochthonous cases of Leishmania (Leishmania) infantum in horse (Equus caballus) in the Americas and mixed infection of Leishmania infantum and Leishmania (Viannia) braziliensis. Vet Parasitol 197:665–669. https://doi.org/10.1016/j.vetpar.2013.06.014
Sousa-Paula L, Pessoa F, Otranto D, Dantas-Torres F (2021) Beyond taxonomy: species complexes in New World phlebotomine sand flies. Med Vet Entomol 35:267–283. https://doi.org/10.1111/mve.12510
Souza NA, Andrade-Coêlho CA, Vilela ML, Rangel EF (2001) The Phlebotominae sand fly (Diptera: Psychodidae) fauna of two atlantic rain forest reserves in the State of Rio de Janeiro, Brazil. Mem Inst Oswaldo Cruz 96:319–324
Taniguchi HH, Tolezano JE, Larosa R, Elias CR, Galati EAB (2002) Sandfly ecological observations in Eldorado County, Ribeira Valley, São Paulo State, Brazil, an endemic area of American Cutaneous Leishmaniasis. Period between 1996 and 1997. I-Sazonality and frequence of Lutzomyia ayrozai to different ecotopes with sent. Rev Inst Adolfo Lutz 61:103–112
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Torrellas A, Ferrer E, Cruz I, Lima H, Delgado O, Rangel JC, Bravo JA, Chicharro C, Llanes-Acevedo IP, Miles MA, Feliciangeli MD (2018) Molecular typing reveals the co-existence of two transmission cycles of american cutaneous leishmaniasis in the andean region of venezuela with Lutzomyia migonei as the vector. Mem Inst Oswaldo Cruz 113:1–8. https://doi.org/10.1590/0074-02760180323
Truppel H, Otomura F, Teodoro U, Massafera R, Costa-Ribeiro MCV, Catarino CM, Dalagrana L, Ferreira MEMC, Thomaz-Soccol V (2014) Can equids be a reservoir of Leishmania braziliensis in endemic areas? PLoS ONE 9:1–6. https://doi.org/10.1371/journal.pone.0093731
Acknowledgements
SLM thanks Daniela Lamattina, Nilso Molina and Milena Casafus from INMeT-ANLIS Malbrán for their collaboration in the field work. Also to Elizabet Vilacoba and Dario Lijtmaer of the Museo Argentino de Ciencias Naturales "Bernardino Rivadavia" for their contributions in the sequencing stage.
Funding
This research was partially supported by Consejo Nacional de Investigacioes Científicas y Técnicas, Argentina, and resources from Instituto Nacional de Medicina Tropical ANLIS “Carlos G. Malbrán”, Ministerio de Salud, Argentina.
Author information
Authors and Affiliations
Contributions
Conceptualization: SLM; methodology: SLM and APM; funding acquisition: SLM and ODS; resources: SLM, MGQ and ODS; supervision: MGQ and ODS; writing original draft: SLM, writing—review & editing: ODS, MGQ, APM and SLM.
Corresponding author
Ethics declarations
Competing Interests
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Edited by Marco Pezzi
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Moya, S.L., Pech-May, A., Quintana, M.G. et al. Cryptic Diversity in Sympatric Migonemyia migonei (Diptera: Psychodidae), Eventual Meaning for Leishmaniasis Transmission. Neotrop Entomol 53, 47–55 (2024). https://doi.org/10.1007/s13744-023-01095-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13744-023-01095-5