
Abstract
In the present study, a new series of 3-(2,5-dichlorothiophen-3-yl)-5-aryl-4,5-dihydro-1H-pyrazole-1-carbothioamides 2 were synthesized either by the reaction of (E)-3-aryl-1-(2,5-dichlorothiophen-3-yl)prop-2-en-1-ones 1 with thiosemicarbazide or by one-pot reaction of 3-acetyl-2,5-dichlorothiophene with the corresponding aldehyde and thiosemicarbazide. Additionally, 2-(3-(2,5-dichlorothiophen-3-yl)-5-aryl-4,5-dihydro-1H-pyrazol-1-yl)-4-phenylthiazoles 3 were synthesized in 46–89% yields by the reflux of carbothioamides 2 with 2-bromoacetophenone. The structures of the newly synthesized compounds were characterized by IR, 1H-NMR, 13C-NMR, DEPT-135, and mass spectrometry analysis (MS). All new compounds were evaluated as antimicrobial and antioxidants. Compound (3b) exhibited moderate activity against Bacillus subtilis and Penicillium fimorum, 14 ± 0.5 mm and 18 ± 0.75 mm, respectively, while the other synthesized compounds did not show activity against the tested microbes. The most potent antioxidant activity showed by compound (2a) and (2e) with 95.2% and 96.3%, which considered good to excellent antioxidant activity compared with the control (ascorbic acid) and other synthesized compounds. Molecular docking study of the new compounds with cytochrome P450 14 alpha-sterol demethylase (CYP51) was carried out to evaluate their possibility as drugs and to implement structural improvements for this purpose. All synthesized compounds exhibited good affinity with (CYP51), notably, (3a) and (3b) compounds showed the highest affinity with the lowest binding energies.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pyrazole bearing carbothioamide moiety are a class of heterocyclic compounds which have shown variety biologically active properties such as anti-inflammatory, analgesic, cytotoxicity against some human tumor cell lines, and the antimicrobial activities [1]. Pyrazole-carbothioamides have been reported with biological properties such as cytotoxicity against Raji cell shown by compound (A), anti-inflammatory activity exhibited by carbothioamide (B), and the antimicrobial activities exerted by compound (C) [1,2,3,4] (Fig. 1).

Examples of biologically active pyrazole-carbothioamide derivatives
Thiazole is a five-membered heterocyclic compound contains nitrogen and sulfur atoms in 1,3-position [5, 6]. Many pyrazoles bearing thiazole substituents showed a remarkable cytotoxic activity, such as compound (D) [7] and compound (E) [8] which exhibited high anticancer activity with good selectivity (Fig. 2).

Examples of biologically active pyrazole-thiazole derivatives
Recently, 3-(2,5-Dichlorothiophen-3-yl)-5-(2,4-dimethoxyphenyl)-1-methyl-4,5-dihydro-1H-pyrazole has been synthesized by our research group [9, 10]. Herein, we report the synthesis, characterization, antioxidant activity, and molecular docking study of 3-(2,5-dichlorothiophen-3-yl)-5-aryl-4,5-dihydro-1H-pyrazole-1-carbothioamides 2 and their thiazole 3 derivatives (Scheme 1).

Synthesis of carbothioamides (2a–f) and thiazoles (3a, 3b, 3d–f)
Experimental
Instrumentation
1H (300 MHz) and 13C (75 MHz) NMR spectra were recorded in DMSO-d6 and CDCl3, which were used as an internal standard at 300 K on Bruker Ultra Shield 300 MHz spectrometer (Bruker, Switzerland). Chemical shifts δ are given in parts per million (ppm) and were determined from the center of the respective coupling patterns (s: singlet, d: doublet, dd: doublet of doublet, t: triplet, q: quartet, pent: pentet, sext: sextet, and m: multiplet). ESIMS measurements were performed on a SHIMADZU gas chromatograph mass spectrometer GCMS-QP5050A (Shimadzu, Japan). The infrared (IR) spectra were obtained using the Nicolet Impact-400 FT-IR spectrophotometer (Thermo-Fisher Scientific). Melting point were determined by the Electrothermal IA 9100 series apparatus (Cole-Parmer). Thin layer chromatography (TLC) monitoring of the reaction was carried out using analytical TLC plates coated with silica gel (60 F254, Merck).
Synthesis
Materials
Solvents were dried and distilled according to standard methods. Chalcones 1a–b, 1d, and 1f were prepared according to the literature [11,12,13,14]; to a methanolic solution containing equimolar amount (1.0 mmol) of aldehydes and KOH, a solution of 3-acetyl-2,5-dichlorothiophene (1.0 mmol) in methanol (10 mL) was added dropwise. The reaction mixture was stirred at room temperature for 10 h. The solid precipitate formed was filtered off, washed with methanol and dried then used without any further purification.
General procedure for the synthesis of carbothioamides (2a-f)
Method A
To a solution of chalcone 1 (1.0 mmol) and potassium hydroxide (2.0 mmol, 2 equiv.) in ethanol (10.0 mL), thiosemicarbazide (2.0 mmol, 2 equiv.) was added with stirring at room temperature. The mixture was refluxed for 5 h and allowed to cool to room temperature. The resulting solid was filtered off, washed with ethanol (2 × 5 mL), and dried to give the corresponding carbothioamides 2a–b, 2d, and 2f as a solid products.
Method B
Potassium hydroxide (4.0 mmol, 4 equiv.) in methanol (5 mL) was added to 50 mL round bottom flask charged with aldehyde 70 (1.0 mmol), 3-acetyl-2,5-dichlorothiophene (1.0 mmol), and thiosemicarbazide (2.0 mmol, 2 equiv.) in methanol (25 mL). The resulting mixture was stirred for 6 h at room temperature followed by reflux for 2–5 h. After cooling to room temperature, the precipitated solid was filtered off, washed with methanol (2 × 5 mL), and dried to afford carbothioamides 2c and 2e as a solid products.
3-(2,5-Dichlorothiophen-3-yl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (2a)
Light brown solid (EtOH); yield (82%); mp 177–178 °C. IR (KBr, cm−1) ν 3399 (N–H), 3151 (C–H), 1636 (C=N), 1346 (C=S). 1H NMR (300 MHz, DMSO-d6): δ 3.13 (d, 1H, 3 J = 17.4 Hz, H-4a), 3.72 (s, 3H, Ph-OCH3), 3.95 (dd, 1H, 2J4a,4b = 17.9 Hz, 3J4b,5 = 11.5 Hz, H-4b), 5.82 (d, 1H, 3 J = 9.5 Hz, H-5), 6.87 (d, 2H, 3 J = 7.9 Hz, 3″,5″-H), 7.04 (d, 2H, 3 J = 8.0 Hz, 2″,6″-H), 7.82 (s, 1H, H-4′), 7.96 (s, 1H, CSNH2), 8.15 (s, 1H, CSNH2). 13C NMR (75 MHz, DMSO-d6): δ 43.8 (C-4), 55.1 (Ph-OCH3), 62.6 (C-5), 113.9 (C-3″, C-5″), 126.0 (C-3′), 126.4 (Cq-5′), 126.6 (C-2″, C-6″), 127.3 (C-4′), 130.1 (Cq-2′), 134.9 (Cq-1″), 148.5 (Cq-3), 158.3 (Cq-4″), 176.3 (CSNH2). EIMS m/z: calc. for C15H13Cl2N3OS2 [M]+ 385; found 385.
3-(2,5-Dichlorothiophen-3-yl)-5-(2-methoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (2b)
Pale yellow solid (EtOH); yield (52%); mp 229–230 °C IR (KBr, cm−1) ν 3433 (N–H), 3127 (C–H), 1635 (C=N), 1347 (C=S). 1H NMR (300 MHz, DMSO-d6): δ 3.03 (d, 1H, 3 J = 18.6 Hz, H-4a), 3.82 (s, 3H, Ph-OCH3), 3.92 (dd, 1H, 2 J 4a,4b = 18.2 Hz, 3 J 4b,5 = 11.5 Hz, H-4b), 6.03 (d, 1H, 3 J = 11.1 Hz, H-5), 6.80–6.89 (m, 2H, 3″,4″-H), 7.03 (d, 1H, 3 J = 8.4 Hz, 6″-H), 7.25 (t, 1H, 3 J = 7.5 Hz, 5″-H), 7.78 (s, 1H, H-4′), 7.93 (s, 1H, CSNH2), 8.09 (s, 1H, CSNH2). 13C NMR (75 MHz, DMSO-d6): δ42.5 (C-4) 55.6 (Ph-OCH3) 59.0 (C-5) 111.4 (C-3″) 120.1 (C-5″) 125.4 (Cq-2′) 125.9 (C-4′) 127.2 (C-6″) 128.3 (C-4″) 129.7 (C-3′) 130.0 (Cq-1″) 149.1 (Cq-3) 155.5 (Cq-2″) 176.4 (CSNH2). EIMS m/z: calc. for C15H14Cl2N3OS2 [M + H]+ 386; found 386.
3-(2,5-Dichlorothiophen-3-yl)-5-(3-methoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (2c)
Yellow solid (MeOH); yield (71%); mp 204–205 °C (Dec.). IR (KBr, cm−1) ν 3416 (N–H), 3076 (C–H), 1613 (C=N), 1358 (C=S). 1H NMR (300 MHz, DMSO-d6): δ 3.13 (d, 1H, 3 J = 17.0 Hz, H-4a), 3.71 (s, 3H, Ph-OCH3), 3.97 (dd, 1H, 2 J 4a,4b = 17.8 Hz, 3 J 4b,5 = 11.5 Hz, H-4b), 5.86 (d, 1H, 3 J = 11.4 Hz, H-5), 6.64 (s, 2H, 2″,4″-H), 6.81 (d, 1H, 3 J = 8.3 Hz, 6″-H), 7.24 (t, 1H, 3 J = 7.9 Hz, 5″-H), 7.80 (s, 1H, H-4′), 8.01 (s, 1H, CSNH2), 8.19 (s, 1H, CSNH2). 13C NMR (75 MHz, DMSO-d6): δ 43.8 (C-4), 55.1 (Ph-OCH3), 63.0 (C-5), 111.4 (C-4″), 112.0 (C-2″), 117.1 (C-6″), 126.1 (Cq-5′), 126.5 (C-3′), 127.3 (C-5″), 130.0 (Cq-2′), 130.1 (C-4′), 144.4 (Cq-1″), 148.6 (Cq-3), 159.4 (Cq-3″), 176.4 (CSNH2). EIMS m/z: calc. for C15H13Cl2N3OS2 [M]+ 385; found 385.
5-(3-Bromo-4-methoxyphenyl)-3-(2,5-dichlorothiophen-3-yl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (2d)
Light yellow solid (EtOH); yield (69%); mp 211–212 °C (Dec.). IR (KBr, cm−1) ν 3451 (N–H), 3151 (C–H), 1596 (C=N), 1347 (C=S). 1H NMR (300 MHz, DMSO-d6): δ 3.17 (d, 1H, 3 J = 18.1 Hz, H-4a), 3.81 (s, 3H, Ph-OCH3), 3.96 (dd, 1H, 2 J 4a,4b = 18.2 Hz, 3 J 4b,5 = 11.7 Hz, H-4b), 5.83 (d, 1H, 3 J = 11.3 Hz, H-5), 7.07 (s, 2H, 5″,6″-H), 7.30 (s, 1H, 2″-H), 7.82 (s, 1H, H-4′), 8.02 (s, 1H, CSNH2), 8.23 (s, 1H, CSNH2). 13C NMR (75 MHz, DMSO-d6): δ 43.7 (C-4), 56.3 (Ph-OCH3), 62.1 (C-5), 110.4 (C-3′), 112.8 (C-5″), 126.0 (C-6″), 126.1 (Cq-5′), 126.6 (Cq-2′), 127.3 (C-2″), 129.9 (C-3″), 130.0 (C-4′), 136.5 (Cq-1″), 148.6 (Cq-3), 154.4 (Cq-4″), 176.3 (CSNH2). EIMS m/z: calc. for C15H12BrCl2N3OS2 [M]+ 465; found 465.
5-(4-(tert-Butyl)phenyl)-3-(2,5-dichlorothiophen-3-yl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (2e)
Light yellow solid (MeOH); yield (88%); mp 224.225 °C. IR (KBr, cm−1) ν 3427 (N–H), 3128 (C–H), 1572 (C=N), 1347 (C=S). 1H NMR (300 MHz, DMSO-d6): δ 1.25 (s, 9H, Ph-C(CH3)3), 3.14 (d, 1H, 3 J = 18.2 Hz, H-4a), 3.96 (dd, 1H, 2 J 4a,4b = 18.1 Hz, 3 J 4b,5 = 11.5 Hz, H-4b), 5.88 (d, 1H, 3 J = 11.3 Hz, H-5), 7.04 (d, 2H, 3 J = 8.2 Hz, 3″,5″-H), 7.33 (d, 2H, 3 J = 8.3 Hz, 2″,6″-H), 7.78 (s, 1H, H-4′), 7.92 (s, 1H, CSNH2), 8.10 (s, 1H, CSNH2). 13C NMR (75 MHz, DMSO-d6): δ 31.1 (Ph-C(CH3)3), 34.1 (Ph-C(CH3)3), 43.7 (C-4), 62.7 (C-5), 124.9 (C-3″, C-5″), 125.2 (C-2″, C-6″), 125.9 (Cq-5′), 126.3 (Cq-2′), 127.2 (C-4′), 129.9 (C-3′), 139.7 (Cq-1″), 148.5 (C-4″), 149.2 (Cq-3), 176.4 (CSNH2). EIMS m/z: calc. for C18H19Cl2N3S2 [M]+ 411; found 411.
3-(2,5-Dichlorothiophen-3-yl)-5-(naphthalen-1-yl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (2f)
Light yellow solid (EtOH); yield (42%); mp 238–239 °C (Dec.). IR (KBr, cm−1) ν 3445 (N–H), 3127 (C–H), 1595 (C=N), 1370 (C=S). 1H NMR (300 MHz, DMSO-d6): δ (ppm) = 3.05 (d, 1H, 3 J = 17.9 Hz, H-4a), 4.24 (dd, 1H, 2 J 4a,4b = 18.0 Hz, 3 J 4b,5 = 11.7 Hz, H-4b), 6.66 (d, 1H, 3 J = 8.9 Hz, H-5), 6.99 (d, 1H, 3 J = 6.7 Hz, 2″-H), 7.45 (t, 1H, 3 J = 7.7 Hz, 7″-H), 7.55–7.64 (m, 2H, 3″,6″-H), 7.84 (s, 1H, H-4′), 7.98 (d, 1H, 3 J = 7.7 Hz, 8″-H), 8.15 (s, 1H, NH), 8.31 (s, 1H, NH).13C NMR (75 MHz, DMSO-d6): δ 43.3 (C-4), 60.6 (C-5), 121.0, 123.4, 125.6, 125.9, 126.0, 126.4, 126.5, 127.3, 127.5, 128.8, 129.0, 130.0 (Ar–C), 133.8 (Cq-3′), 137.6 (Cq-1″), 149.2 (Cq-3), 176.5 (CSNH2). EIMS m/z:calc. for C18H13Cl2N3S2 [M]+ 405; found 405.
General procedure for the synthesis of thiazoles (3a, 3b, 3d-f)
A solution of carbothioamide 2 (1.0 mmol) and 2-bromoacetophenone (1.0 mmol) in ethanol (10.0 mL) was refluxed for 4–8 h. The resulting mixture was allowed to cool to room temperature, and the resulting solid was filtered off, washed with methanol (2 × 5 mL), and dried to give thiazoles 3.
2-(3-(2,5-Dichlorothiophen-3-yl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-phenylthiazole (3a)
Greenish yellow solid (EtOH); yield (88%); mp 136–137 °C. IR (KBr, cm−1) ν 2926 (C–H), 1543 (C=N). 1H NMR (300 MHz, CDCl3): δ 3.49 (dd, 1H, 2 J 4a,4b = 18.1 Hz, 3 J 4a,5 = 6.4 Hz, H-4a), 3.79 (s, 3H, Ph-OCH3), 4.01 (dd, 1H, 2 J 4a,4b = 18.0 Hz, 3 J 4b,5 = 12.0 Hz, H-4b), 5.76 (d, 1H, 3 J = 3.4 Hz, H-5), 6.81 (s, 1H, 5″′′-H), 6.89 (d, 2H, 3 J = 8.6 Hz, 3″,5″-H), 7.30–7.37 (m, 6H, Ar–H), 7.69 (d, 2H, 3 J = 7. 113.5 (Cq-3′)2 Hz, 2″″,6″″-H). 13C NMR (75 MHz, CDCl3): δ 44.5 (C-4), 54.4 (Ph-OCH3), 63.9 (C-5), 102.3 (C-5″′), 113.6 (C-3″, C-5″), 124.9, 125.7, 127.08, 127.15, 127.2, 127.7, 127.9, 132.0 (Ar–C). EIMS m/z: calc. for C23H18Cl2N3OS2 [M + H]+ 486; found 486.
2-(3-(2,5-Dichlorothiophen-3-yl)-5-(2-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-phenylthiazole (3b)
Pale yellow solid (EtOH); yield (46%); mp 197–198 °C. IR (KBr, cm−1) ν 2966 (C–H), 1613 (C=N). 1H NMR (300 MHz, CDCl3): δ 3.48 (dd, 1H, 2 J 4a,4b = 18.1 Hz, 3 J 4a,5 = 6.4 Hz, H-4a), 3.78 (s, 3H, Ph-OCH3), 4.03 (dd, 1H, 2 J 4a,4b = 18.1 Hz, 3 J 4b,5 = 12.0 Hz, H-4b), 5.77 (d, 1H, 3 J = 3.4 Hz, H-5), 6.81–6.88 (m, 2H, 5″-H, 5″′-H), 7.25–7.70 (m, 9H, Ar–H). 13C NMR (75 MHz, CDCl3): δ 44.5 (C-4), 54.5 (Ph-OCH3), 64.0 (C-5), 102.5 (C-5″′), 113.6 (C-3″, C-5″), 125.9, 126.1, 127.0, 127.1, 127.2, 127.8, 128.1, 133.1 (Ar–C). EIMS m/z: calc. for C23H18Cl2N3OS2 [M + H]+ 486; found 486.
2-(5-(3-Bromo-4-methoxyphenyl)-3-(2,5-dichlorothiophen-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-phenylthiazole (3d)
Yellow solid (EtOH); yield (89%); mp 189–190 °C (Dec.). IR (KBr, cm−1) ν 2927 (C–H), 1549 (C = N). 1H NMR (300 MHz, CDCl3): δ 3.48 (dd, 1H, 2 J 4a,4b = 18.1 Hz, 3 J 4a,5 = 7.1 Hz, H-4a), 3.88 (s, 3H, Ph-OCH3), 4.01 (dd, 1H, 2 J 4a,4b = 18.1 Hz, 3 J 4b,5 = 12.2 Hz, H-4b), 5.62 (dd, 1H, 3 J 4a,5 = 11.6 Hz, 3 J 4b,5 = 7.0 Hz, H-5), 6.83 (s, 1H, 5″′-H), 6.88 (d, 1H, 3 J = 8.5 Hz, 5″-H), 7.23–7.35 (m, 5H, Ar–H), 7.65 (s, 1H, 2″-H), 7.70 (d, 2H, 3 J = 7.3 Hz, 2″″,6″″-H). 13C NMR (75 MHz, CDCl3): δ 45.0 (C-4), 56.4 (Ph-OCH3), 64.3 (C-5), 103.7 (C-5″′), 112.2 (C-3″), 112.4 (Cq-3′), 125.6, 126.0, 126.5, 126.7, 127.5, 127.6, 128.3, 128.6, 128.8, 130.8, 131.7, 131.8 (Ar–C), 156.0 (Cq-4″), 164.8 (C-2″′). EIMS m/z: calc. for C22H14BrCl2N3OS2 [M + H-CH3]+ 551; found 551.
2-(5-(4-(tert-Butyl)phenyl)-3-(2,5-dichlorothiophen-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-phenylthiazole (3e)
Yellow solid (EtOH); yield (74%); mp 179–180 °C. IR (KBr, cm−1) ν 2955 (C–H), 1544 (C=N). 1H NMR (300 MHz, CDCl3): δ 1.30 (s, 9H, Ph-C(CH3)3), 3.51 (dd, 1H, 2 J 4a,4b = 17.9 Hz, 3 J 4a,5 = 6.8 Hz, H-4a), 4.00 (dd, 1H, 2 J 4a,4b = 18.0 Hz, 3 J 4b,5 = 12.0 Hz, H-4b), 5.74 (d, 1H, H-5), 6.81 (s, 1H, 5″′-H), 7.29–7.40 (m, 8H, Ar–H), 7.66 (d, 2H, 3 J = 7.2 Hz, 2″″,6″″-H). 13C NMR (75 MHz, CDCl3): δ 31.40 (Ph-C(CH3)3), 31.44 (Ph-C(CH3)3), 45.5 (C-4), 65.1 (C-5), 103.3 (C-5″′), 125.9, 126.1, 126.2, 126.4, 126.7, 127.5, 127.8, 128.15, 128.21, 128.3, 128.7, 128.8, 129.1, 129.7 (Ar–C). EIMS m/z: calc. for C26H24Cl2N3S2 [M + H + 2]+ 514; found 514.
2-(3-(2,5-Dichlorothiophen-3-yl)-5-(naphthalen-1-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-phenylthiazole (3f)
Greenish yellow solid (EtOH); yield (72%); mp 186–187 °C. IR (KBr, cm−1) ν 3053 (C–H), 1549 (C=N). 1H NMR (300 MHz, CDCl3): δ 3.48 (dd, 1H, 2 J 4a,4b = 17.6 Hz, 3 J 4a,5 = 6.8 Hz, H-4a), 4.20 (dd, 1H, 2 J 4a,4b = 17.9 Hz, 3 J 4b,5 = 12.2 Hz, H-4b), 6.50 (s, 1H, H-5), 6.84 (s, 1H, 5″′-H), 7.18–8.10 (m, 13H, Ar–H). 13C NMR (75 MHz, CDCl3): δ 45.1 (C-4), 63.3 (C-5), 103.5 (C-5″′), 106.5 (Cq-3′), 110.7, 113.3, 113.7, 114.3, 114.7, 117.3, 117.6, 119.0, 120.6, 121.0, 121.4, 123.5, 124.6, 125.0, 125.5, 126.0, 126.1, 126.4, 126.8, 127.4, 127.8, 128.1, 128.7, 129.3 (Ar–C).
Antioxidant activity assay
1,1-Diphenyl-2-picrylhydrazyl (DPPH) assay was carried out to determine the antioxidant activity of thioamides (2a-f) or thiazoles (3a, 3b, 3d-f). Briefly, 3.94 mg of DPPH were dissolved in 100 mL methanol to acquire (0.1 mM) stock solution. Pyrazole or thiazole were prepared in a concentration of 4.0 mg/mL dissolved in DMSO. A 2 mL solution of thioamides (2a-f) or thiazoles (3a, 3b, 3d-f) was mixed with 3 mL of DPPH and allowed to stand in dark at room temperature. DPPH absorbance was measured at 517 nm after 30 min and 60 min using a spectrophotometer [15]. The following equation was used to calculate the antioxidant activity:
Abs sample = absorbance of the sample with DPPH; DPPH = absorbance of DPPH. The antioxidant activity of the tested compounds was compared to ascorbic acid which was used as a positive control.
To determine EC50 (50% radical scavenging activity) values, one of each group was chosen (2a and 3a), solutions of concentrations (1, 2, 4, 8, and 16 µmol/mL) of 2a and (0.8, 1.6, 3.2, 6.4, and 12.8 µmol/mL) of 3a were freshly prepared. Antioxidant activity was determined by mixing 2 mL of each solution of 2a or 3a with 3.0 mL of DPPH and allowed to stand in dark for 30, 60, 120, and 180 min, at room temperature. The absorbance measured at 517 nm were plotted against sample concentration, and EC50 values were determined from the graphs.
Molecular docking study and drugability
The process of classifying compounds as drugs depends on several criteria. An integrated system like the Biopharmaceutics Drug Disposition Classification System (BDDCS) that adopted the Lipinski’s Rule of 5 to appoint the drugability of these compounds [16]. According to Lipinski's Rule of 5, the ability of a compound to be a drug and to increase its absorption has limitations such as MW (≤ 500 g/mol), H-bond donors (≤ 5), H-bond acceptors, (≤ 10), n-octanol/water partition coefficient (Log Po/w ≤ 5) [17], predicted by using SwissADME website (SwissADME, www.swissadme.ch, accessed on June.2021), in addition to the inhibiting property of many types of CYP-Protein systems [18].
In this study, The cytochrome P45014 alpha-sterol demethylase (CYP51) complexed with fluconazole was selected for docking study and downloaded from the protein databank (PDB ID: 1EA1) [19], and the structure of the isoniazid as an experimental control was downloaded from PubChem database (PubChem CID: 3767) then converted from sdf format to PDB (https://pubchem.ncbi.nlm.nih.gov/compound/Isoniazid, accessed on May.2021). 2D structure of carbothioamides (2a–f) and thiazoles (3a, 3b, 3d–f) were drawn in ChemDraw Professional 16.0 and subjected to energy minimization by using Chem3D 16.0, then the sdf format converted to PDB.
The AutoDock preparation tools-1.5.6 were used to prepare PDBs for docking process by removing water molecules and any atoms that unrelated to the protein from the protein complex, and adding the missing hydrogen atoms and the kollman charges. After receptor and inhibitor separation from the crystal complex and structures minimization [20], the binding sites in CYP51 were recognized for setting the grid size to be − 18, − 2.6, and 64 for X, Y, and Z dimensions, respectively, with a grid center 78, 76, and 84 along the X, Y, and Z dimensions, respectively, with a grid spacing equal to 0.375 Å, according to the literature procedure. The possible binding affinities and energies were then calculated by the AutoDock Vina (version 1.1.2, Scripps Research Institute, La Jolla, CA, USA). [21]. The interactions were analyzed by Biovia Discovery Studio Visualizer Client 2021 (Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2021).
Results and discussion
Chemistry
The chalcones (1a-b, 1d, and 1f) was treated with thiosemicarbazide in the presence of potassium hydroxide using ethanol as a solvent. The reaction lead to the formation of the racemic pyrazole derivatives (2a-b, 2d, and 2f) bearing carbothioamide subunits. Carbothioamides (2a and 2e) produced through one-pot reaction of 3-acetyl-2,5-dichlorothiophene with the corresponding aldehydes in the presence of thiosemicarbazide and potassium hydroxide using methanol as a solvent. All produced carbothioamides were treated later with 2-bromoacetophenone to produce the desired thiazoles 3 in 46–89% yields (Scheme 1, Table 1).
The newly prepared thioamides (2a–f) and thiazoles (3a, 3b, 3d–f) were characterized using different spectroscopic methods including IR, 1H-NMR, 13C-NMR, and mass spectrometry. Complete assignments for all protons and carbons are given in the experimental part. The ESIMS data analyses revealed the correct molecular ion peaks for all compounds as suggested by their molecular formulas.
Infrared spectra of new carbothioamides (2a–f) showed all expected functional groups in the new pyrazole derivatives. In the IR spectra of the carbothioamides, the main absorption bands observed at 3399–3451 cm−1, 3076–3151 cm−1, 1595–1636 cm−1, and 1346–1370 cm−1 can be attributed for N–H, C–H, C=N, and C=S vibrations, respectively. The infrared spectra of the thiazoles (3a, 3b, 3d–f) showed the main absorption bands observed at 3053–2966 cm−1 and at 1543–1613 cm−1 may be attributed to the C–H and C=N vibrations, respectively. The absence of N–H and C=S absorption bands indicates the formation of thiazoles 3.
The 1H NMR spectra of all new carbothioamides (2a–f) showed two geminal protons appeared as a doublet and doublet of doublet, observed at δ 3.03–3.17 ppm and δ 3.92–4.24 ppm attributed to H-4a and H-4b, respectively. While, the vicinal proton H-5 observed as a doublet at δ 5.82–6.66 ppm. The thiophene ring proton peak appeared as a singlet at δ 7.78–7.84 ppm. The two singlet peaks at δ 7.92–8.15 ppm and δ 8.09–8.31 ppm assigned to the protons of the thioamide group.
The 1H-NMR spectra of thiazoles (3a, 3b, 3d–f) exhibit two geminal protons appeared as a doublet of doublet at δ 3.48–3.51 and δ 4.00–4.20 for H-4a and H-4b, respectively. The H-5 proton observed at δ 5.74–6.50. A singlet peak detected at δ 6.81–6.84 assigned to thiazole ring proton and the absence of the peaks of the NH2 protons proved the formation of thiazoles 3.
The 13C-NMR spectra of carbothioamides (2a–f) showed two signals at δ 42.5–43.8 and δ 59.0–63.0 attributed to C-4 and C-5 carbons of the pyrazoline ring, respectively. The signal appeared at δ 176.3–176.5 assigned to C=S carbon. The 13C-NMR spectra of thiazoles (3a, 3b, 3d–f) showed a signal at δ 102.3–103.7 attributed to CH carbon of the thiazole ring, while the absence of C=S signal in the final product proved the formation of thiazoles 3.
Antioxidant activity test
Antioxidant activities of racemic thioamides (2a–f) and thiazoles (3a, 3b, 3d–f) were measured using DPPH with concentration of (4.0 mg/mL); Fig. 3. All compounds (2a–f) and (3a, 3b, 3d–f) showed moderate to excellent antioxidant activity compared to ascorbic acid (control), with compounds (2a) and (2e) being the highest active with a scavenging percentage of 95.2% and 96.3%, respectively. Notably, the activity of all pyrazole bearing carbothioamide subunit (2a–f) was higher than the activity of thiazoles (3a, 3b, 3d–f).

The DPPH radical scavenging activity of (2a–f) and (3a, 3b, 3d–f) compounds
DPPH is one of the free radicals widely used for testing antioxidant activity of a chemical compounds or a plant extracts. In the present study, pyrazoles (2a) and thiazoles (3a) were showed potential free-radical scavenging activity at different concentrations, Figs. 4 and 5, respectively. The antioxidant activities of the individual compounds, may depend on structural factors, such as the number of phenolic hydroxyl or methoxy groups, flavone hydroxyl, keto groups, free carboxylic groups, and other structural features [22, 23].

The DPPH radical scavenging activity of 2a at different concentrations and incubation times

The DPPH radical scavenging activity of 3a at different concentrations and incubation times
From the Figs. 4 and 5, EC50 of pyrazole (2a) and thiazole (3a) were calculated, it is found that EC50 of thioamide (2a) and thiazole (3a) were 3.78 and 2.85 μmol/mL, respectively, after 180 min. Lower EC50 values indicate higher radical scavenging potential. Additionally, there was a very small changes in antioxidant activity of all samples with time (Table 2). The antioxidant activity may be due to the presence of methoxy, carbothioamide, and thiazole moieties.
Molecular docking simulation
As shown in Table 3, all synthesized compounds match the rules of Lipinski studied, except the molecular weight of compounds 3d, 3e, and 3f. The lipophilicity plays an essential role in drugability criteria and expressed by the n-octanol/water partition coefficient (Log Po/w) values. For the synthesized compounds, the partition coefficient ranged between 3.02–3.63 and between 4.13–4.77 for carbothioamides 2 and thiazoles 3, respectively. Additionally, the numbers of H-bond donors and acceptors were in good agreement with the Lipinski’s rules.
Cancer cells multiply and producing cholesterol to build their membranes. Sterol 14α-demethylase (CYP51) is potentially a more specific target for anticancer chemotherapy, because it’s fully committed to cholesterol production. So, the molecular docking was performed on this protein [24]. Moreover, previous studies pointed out that sterol 14α-demethylase (CYP51) is a target of azole drugs [25]. The cytochrome P45014 alpha-sterol demethylase (CYP51) complexed with fluconazole was downloaded from the protein databank (PDB ID: 1EA1) and used as a reference of docking process. The structure of the isoniazid as an experimental control was downloaded from PubChem database (PubChem CID: 3767) then converted from sdf format to PDB.
After preparation of protein, ligands and derivatives for docking by AutoDock preparation tools-1.5.6, AutoDock Vina 1.1.2 were used to calculate the possible binding sites and energies between CYP51 and the controls (flucaonazole and isoniazide) of our derivatives 2 and 3 (Table 3).
The binding energy values for all pyrazole bearing carbothioamides 2 and thiazoles 3 were higher than the experimental control (isoniazid), and some were even higher than the control inhibitor (fluconazole). Thiazoles 3 showed lower binding energy compared to carbothioamides 2. It is noteworthy that thiazoles bearing methoxy group on para and ortho positions such as (3a) and (3b) showed the lowest binding energies values of -9.2 and -9.6, respectively, (Table 3).
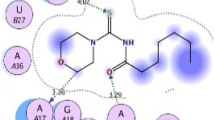
The interactions of racemic carbothioamides 2 and thiazoles 3 with CYP51 are shown in Fig. 6. The carbothioamide (2a) docked inside active site 1 and demonstrated hydrogen bond interactions between thioamide group with Ala256, Thr260, and Cys394, whereas the Pi-Pi T-shaped interaction established between thiophene group with Phe399. Six hydrophobic interactions formed with Met79, Phe83, Ala104, Leu105, Leu152, and Ala256.

3D representation of carbothioamides 2 and thiazoles 3 showing their interactions with the CYP51 active sites. The interaction modes of the highest docking score in CYP51: Pi-cation/Pi-anion interaction (orange), Pi-sulfur (light brown), conventional hydrogen bond interaction (green), Pi-Donor hydrogen bond/carbon hydrogen bond interactions (light blue), alkyl/Pi-alkyl interactions (light purple), Pi-sigma interaction (purple), and Pi-Pi T-shaped/Pi-Pi Stacked interaction (pink)
The interactions between CYP51 and compound (2b) in active site 2 demonstrates hydrogen bond between pyrazole nitrogen and thioamide group with Arg354, and weak Pi-Donor hydrogen bond illustrated phenyl interacted to His318. Two Pi-Cation interactions between thiophene group and phenyl with Arg354 and His430, respectively. In addition to four hydrophobic interactions with Ile27, Ile351, Arg354, and Tyr426. Whereas the compound (2c) demonstrated three hydrogen bond interactions between methoxy group with Lys155, and thioamide group with Ala150 and Cys151. Four hydrophobic interactions established with Ala103, Ala104, Lys155, and Lys156 inside active site 3. Carbothioamide (2d) also docked inside active site 1 by hydrophobic interactions with Phe78, Met79, Phe83, Phe255, His259, Leu315, Pro320, Leu321, Phe387, Cys394, and Ala400 demonstrated.
In the active site 4, only one hydrogen bond existed for carbothioamide (2e) between thioamide group with Thr80. In addition to five hydrophobic interactions with Lys48, Lys74, Pro77, Tyr181, and Leu344. Unfavorable Donor-Donor and hydrogen bond interactions between thioamide group with Asn428 illustrated when the compound (2f) docked inside active site 2 of CYP51. Pi-Pi T-shaped and Pi-Cation interactions between naphthalene group with His430 are present along with Pi-Cation interaction between thiophene group with Arg354. Hydrophobic interactions with Ile27, Leu317, Arg354, Arg427, Asn428, and His430 were demonstrated, in addition to Pi-sulfur interaction between thioamide group and His318 and His430.
Only one Pi-Pi T-shaped interaction illustrated between thiophene group with Phe387 when the thiazole (3a) docked inside active site 1. Many hydrophobic interactions formed with Met79, Arg96, Pro320, Phe387, Cys394, Val395, and Ala400.
The interactions between CYP51 and compound (3b) in the active site 1 demonstrates only hydrophobic interactions with Leu100, Phe255, Ala256, His259, Leu321, Leu324, and Cys394. Thiazole (3d) docked in the active site 2, and demonstrates hydrogen bond interaction between pyrazoline ring with Arg274. Two other active sites show hydrophobic interactions between thiophene group with His318, and His430, and between thiazole group with Glu271, and Arg274, in addition to Pi-cation interaction between thiazole ring with Arg274 when docked with CYP51.
Thiazole (3e) also docked inside active site 2 of CYP51. Only one Pi-Pi Stacked interaction illustrated between thiophene group with Trp267, and two Pi-cation interactions between Arg274 with thiophene and thiazole rings. Additionally, Trp267, Arg274, His363, and Pro423 were interacted via hydrophobic interactions. Two Pi-cation interactions formed between thiophene and thiazolyl phenyl groups with Arg274 and His430, respectively, when the compound (3f) docked in the active site 2. A lot of hydrophobic interactions were indicated with Ile27, His318, Pro319, and Ala350, in addition to Pi-Donor interaction with His318 (Fig. 6).
Conclusion
A new racemic 3-(2,5-Dichlorothiophen-3-yl)-5-arylpyrazole-1-carbothioamides 2 and their thiazole derivatives 3 have been synthesized and characterized using different spectroscopic techniques. The new racemate compounds were evaluated for their antimicrobial and antioxidant activities. Even that, the tested compounds 2 and 3 showed weak antimicrobial activity toward all bacteria and fungi used in this study, they possess a remarkable antioxidant activity compared to ascorbic acid (control). Notably, the activity of all pyrazole bearing carbothioamide subunit (2a–f) was higher than the activity of thiazoles (3a, 3b, 3d–f).
References
K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. Mabkhot, F. Al-aizari, M. Ansar, Molecules 23, 134 (2018)
S.-L. Zhu, Y. Wu, C.-J. Liu, C.-Y. Wei, J.-C. Tao, H.-M. Liu, Bioorg. Med. Chem. Lett. 23, 1343 (2013)
S. Hussain, D. Kaushik, J. Saudi Chem. Soc. 19, 274 (2015)
R. Nagamallu, B. Srinivasan, M.B. Ningappa, A.K. Kariyappa, Bioorg. Med. Chem. Lett. 26, 690 (2016)
A. Taurins, J.G.E. Fenyes, R.N. Jones, Can. J. Chem. 35, 423 (1957)
N.M. Aljamali, J. Plast. Polym. 1, 49 (2015)
Y.H. Zaki, M.S. Al-Gendey, A.O. Abdelhamid, Chem. Cent. J. 12, 70 (2018)
Z. Lin, Z. Wang, X. Zhou, M. Zhang, D. Gao, L. Zhang, P. Wang, Y. Chen, Y. Lin, B. Zhao, J. Miao, F. Kong, Cell Death Dis. 11, 551 (2020)
M.M. Ibrahim, M. Al-Refai, K. Ayub, B.F. Ali, J. Fluoresc. 26, 1447 (2016)
M.M. Ibrahim, M. Al-Refai, B.F. Ali, A. Geyer, K. Harms, M. Marsch, IUCrData 4, x191046 (2019)
M.M. Ibrahim, M. Al-Refai, R. Abu-El-Halawa, H. Tashtoush, S. Alsohaili, M. Masad, Jordan J. Chem. 7, 115 (2012)
M. Al-Refai, M.M. Ibrahim, S. Alsohaili, A. Geyer, Phosph. Sulfur Silicon Relat. Elem. 192, 560 (2017)
M. Al-Refai, M. Ibrahim, A. Al-Fawwaz, A. Geyer, Eur. J. Chem. 9, 375 (2018)
M. Al-Refai, M.M. Ibrahim, M.N. Azmi, H. Osman, M.H. Bakar, A. Geyer, Molecules 24, 4072 (2019)
R. Mothana, U. Lindequist, R. Gruenert et al., Pharmazie 64, 260 (2009)
L.Z. Benet, C.M. Hosey, O. Ursu, T.I. Oprea, Adv. Drug Deliv. Rev. 101, 89 (2016)
C.-Y. Wu, L.Z. Benet, Pharm. Res. 22, 11 (2005)
A. Daina, O. Michielin, V. Zoete, Sci. Rep. 7, 42717 (2017)
L.M. Podust, T.L. Poulos, M.R. Waterman, Proc. Natl. Acad. Sci. 98, 3068 (2001)
J. Wang, W. Wang, P.A. Kollman, D.A. Case, J. Mol. Graph. Model. 25, 247 (2006)
O. Trott, A.J. Olson, J. Comput. Chem. 31, 455 (2009)
D. E. Pratt and B. J. F. Hudson, in Food Antioxid., edited by B. J. F. Hudson (Springer Netherlands, Dordrecht, 1990), pp. 171–191.
M.A.R. Bhuiyan, M.Z. Hoque, S.J. Hossain, World. J. Agric. Sci. 5, 318 (2009)
T.Y. Hargrove, L. Friggeri, Z. Wawrzak, S. Sivakumaran, E.M. Yazlovitskaya, S.W. Hiebert, F.P. Guengerich, M.R. Waterman, G.I. Lepesheva, J. Lipid Res. 57, 1552 (2016)
J. Zhang, L. Li, Q. Lv, L. Yan, Y. Wang, Y. Jiang, Front. Microbiol. 10, 691 (2019)
Acknowledgements
We are grateful to Al al-Bayt University (Mafraq, Jordan) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ibrahim, M.M., Abumahmoud, H. & Al-Fawwaz, A.T. Synthesis, characterization, antimicrobial, antioxidant, and molecular docking study of 3-(2,5-dichlorothiophen-3-yl)-5-arylpyrazole-1-carbothioamides and their thiazole derivatives. J IRAN CHEM SOC 19, 2811–2822 (2022). https://doi.org/10.1007/s13738-022-02495-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-022-02495-x