
Abstract
A thermal reduction is a promising approach for the synthesis of graphene owing to its eco-friendly nature, cost-effective and mass production. However, the elucidation of the structural conformation in graphene oxide concerning for the reduction in temperature remains unclear. In this study, commercially available graphite (Gr) powder was first exploited in distilled water, which was chemically oxidized by using sulphuric acid, potassium permanganate and sodium nitrate. The chemically reduced graphene oxide (GO) was further reduced in a tubular furnace under an inert atmosphere. The Fourier transform infrared spectroscopy analysis revealed the presence of carbonyls, hydroxyls, ethers, epoxides and ketones present between the stacked layers of graphene oxide, which were eliminated in the thermally reduced graphene oxide (rGO) at the temperature range of 100–600 °C. The atomic concentration of oxygen and carbon in the graphene oxide and reduced graphene oxide were evaluated from the X-ray photoelectron spectroscopy. The atomic concentration of oxygen in graphene oxide was 29.80% (w/w) that decreased to 13.68% at 500 °C reduction temperature, confirming the elimination of oxygen-containing functional groups from the stacked structure of graphene oxide. The purity of graphene obtained from the thermal reduction of graphene oxide was 87% which has never been reported earlier.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Graphene is a two-dimensional thin layer of sp2 bonded carbon atoms with some scarce properties like outstanding mechanical rigidity, strength, elasticity, exceptional electrical and thermal conductivity, etc. Since the exploration of graphene in 1947, it has gained vast attention of researchers due to its massive potential application in transistors, sensors, transparent conductive films, catalyst and electrodes because of its excellent electronic, optical, thermal and mechanical performance [1,2,3]. Numerous methods have been developed for the synthesis of graphene such as micro-mechanical exfoliation of bulk graphite [4], epitaxial growth [5], chemical vapour deposition [6] and reduction of graphene oxide [7, 8]. Among these methods, micro-mechanical exfoliation, epitaxial growth and chemical vapour deposition produce high-quality graphene, but their application is limited because of the low yield of graphene and high production cost [9,10,11,12].
The reduction of GO for the synthesis of graphene has gained massive interest amongst researchers due to the large-scale production [13], cost-effectiveness and scalability of the process [14]. The existence of carbonyl and carboxyl groups at the sheet edges of sp2 region along with the presence of hydroxyl and epoxy groups on sp3-hybridized carbon basal plane makes the GO highly hydrophilic in nature [15]. Due to its high hydrophilicity, GO can be easily exfoliated in water through sonication and exfoliated GO can be reduced into graphene by thermal [16], mechanical exfoliation [17] or chemical reduction methodologies such as sodium borohydride [18], hydrazine [19], N,N-dimethylformamide [20], hydroquinone [21], ascorbic acid [22], 1,1-dimethylhydrazine [23] or metal assisted reduction methodology [16]. However, time-consuming reduction time, low yield, higher reduction temperature and deficient reduction of oxygen-containing functional groups limit the applicability of chemical reduction method for the synthesis of graphene from GO. In addition, the reductants used in chemical reduction method are highly lethal and harmful to the human body [24, 25]. Obtained defective nature, production of GO gave significant desirability due to the attraction of various functional groups and incomplete removal of numerous oxygen functional groups [18, 26]
Despite this, to attenuate the challenges faced by chemical reduction method, an alternative method for the reduction of GO can be considered. Thermal reduction route is a viable approach for the reduction of GO due to its eco-friendly nature, high yield and purity of graphene obtained. Thermal reduction of GO removes oxygen-containing functional groups through a series of reactions and releases CO2 and CO from the carbon lattice structure [16, 27, 28]. The graphene acquired from the thermal reduction method exhibits unique physical, chemical and mechanical properties depending upon the reduction temperature [29, 30]. But, the thermal reduction of GO is a very complex because of the multistep removal processes of interpolated H2O molecules and oxygen-containing functional groups carboxyl group, hydroxyl group and epoxy group [31,32,33]. The varied range of oxygen-containing functional groups on the periphery of GO and rGO material makes it to easily exfoliate and functionalized to produce well-dispersed sheet in both water and organic solvent like that in nanocomposites [34] and battery [35]. However, at the high thermal reduction temperature, H2 acts as a reducing agent to mitigate released O2 that is released from GO material during the reduction process [36]. Gao et al. also reported hydroxyl and epoxy groups attached to the inner aromatic domains and they are easily removed from the lattice structure of GO [37]. Therefore, the thermal reduction of GO and resulting graphene needs to be studied in detail.
In the present study, GO was synthesized using modified Hummers method from the commercially available graphite powder. Further GO was reduced thermally to obtain rGO by varying the reduction temperature from 100 to 600 °C. Up to 75%, pour rGO material was acquired by the thermal reduction of GO from 100 to 200 °C. Despite this, by the growth of reduction temperature from 300 to 600 °C we have seen drastic changes, and purity of rGO material was obtained up to 85–87% by the thermal reduction of GO which has been not ever reported earlier. The changes in the structural morphology of GO during the thermal reduction were analysed from X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), atomic force microscope (AFM), Raman analysis and X-ray photoelectron spectroscopy (XPS).
Materials and methods
Chemicals and reagents
Graphite (98%, w/w, mesh size: 60) and hydrogen peroxide (30%, w/v) were obtained from Loba Chemie Pvt Ltd (Mumbai, India). Sodium nitrate (99.5%, w/w) and hydrochloric acid (35%, w/v) were purchased from S D Fine-chem Ltd (Mumbai, India). Potassium permanganate (98.5%, w/w) and sulphuric acid (97.0%, v/v) were procured from Merck Specialities Pvt Ltd (Mumbai, India). All the chemicals were of analytical grade.
Preparation of graphene oxide
GO was synthesized from the natural graphite powder by using modified Hummer’s method [38]. 2.0 g of graphite powder, 1.0 g of NaNO3 and 46 ml of H2SO4 were taken in a volumetric flask (500 ml). After that, KMnO4 (6.0 g) was added slowly under constant stirring at 5 °C for 30 min and 92 ml of distilled water was further added dropwise to the solution at 35 °C. After 30 min, 80 ml distilled water was added and the solution was kept at 98 °C. A light yellow homogenous suspension was obtained after 30 min. Then, 25 ml of H2O2 (30%, w/v) was slowly added to the suspension to reduce the residual KMnO4 and the suspension was kept overnight. Finally, the slurry solution was centrifuged at 9000 rpm, which was further washed with HCl (5%, v/v) to remove metal ions, proceeded by washing with distilled water until the solution pH reached 7.
Preparation of reduced graphene oxide
The reduced graphene oxide (rGO) was prepared from the thermal reduction of GO in a tubular furnace under the nitrogen atmosphere. The GO (100 mg) was taken in a crucible and kept in a tubular furnace in the presence of nitrogen atmosphere. The reduction of GO was carried out for 30 min at different temperatures ranging from 100 to 600 °C. The thermal reduction of GO to rGO was carried out for 30 min at different temperatures ranging from 100 to 600 °C with a ramp rate of 8 °C min−1.
Characterization techniques
Characterization of Gr, GO and rGO was done by using X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM) and Raman spectroscopy analysis. FTIR spectra of the samples were achieved to compare the active surface functional groups present in Gr, GO and rGO with FTIR instrument (PerkinElmer, model: RX-I, Tokyo, Japan) using KBr pellet. The spectra were recorded with a resolution of 1 cm−1 and wave number range between 4000 and 400 cm−1. X-ray diffraction patterns of the Gr, GO and rGO were analysed by X-ray powder diffractometer (Philips, X’PERT-PRO PW3064, Tokyo, Japan) using Cu-K-α radiations (λ = 1.54 Å). The particle size and interplanar spacing between the atoms (d-spacing) were calculated from \(D = 0.94\lambda /\beta \sin \theta\) and \(n\lambda = 2d\sin \theta\), where ‘λ’ was wavelength of X-ray (0.1541 nm), ‘β’ was FWHM (full width at half maximum), ‘θ’ was the diffraction angle, ‘D’ was particle diameter size and ‘d’ was the interplanar spacing between the atoms. The chemical arrangement and bonding state of Gr, GO and rGO were identified from X-ray photoelectron spectroscopy by means of PHI5000 Versa probe II FEI Inc system (Physical Electronics, USA) with monochromatic Al Kα radiation (1486.6 eV). Raman spectroscopy of the procured Gr, GO and rGO before and after thermal treatment was analysed by monochromatic laser source using laboratory RAM HR evolution (Kyoto, Japan) with 532 nm excitation energy. Atomic force microscopy on non-contact mode was used to analyse the surface morphology and average particle size of Gr, GO and rGO nanoparticles (Multimode 8-HR, Massachusetts, USA).
Results and discussion
XRD analysis
The crystal structure, lattice parameter and phase purity of Gr, GO and rGO were examined from XRD analysis and are shown in Fig. 1. The XRD diffractogram of Gr showed a reflection peak at 26.42° (2θ) corresponding to [002] crystal plane and 0.337 nm interlayer spacing [39]. After the oxidation of Gr to GO, two reflection peaks appeared at 10.70° (2θ) and 42.06° (2θ) conforming to [001] and [100] crystal planes, respectively. The peak at 26.42° (2θ) disappeared after the oxidation of Gr indicating the formation of chemically oxidized GO [40,41,42]. It can be seen that the XRD patterns of rGO-100 and GO were almost similar. As the reduction temperature was increased from 100 to 200 °C, the peak corresponding to [001] crystal plane disappeared and a weak broad diffused peak appeared at 24.54° (2θ) [43]. The appearance of new peak in the rGO-200 diffractogram was due to the presence of oxygen and moisture content stacked between the layers of rGO sheet. Babak et al. [44] also observed the appearance of new peak at 24° (2θ) due to the presence of oxygen and moisture content. With further increase in the reduction temperature from 200 to 600 °C, the characteristic peak of rGO shifted to 25.85° (2θ) conforming to [002] crystal plane [45].

X-ray diffraction of graphite, chemically reduced graphene oxide and thermally reduced graphene oxide at different temperatures
The interlayer spacing (d-spacing), crystalline size and lattice strain were calculated from XRD analysis of Gr, GO and rGO and are given in Table 1. The d-spacing for Gr was 0.337 nm that increased to 0.826 nm after the chemical oxidation of Gr into GO. The increase in the interlayer d-spacing was due to the entrapping of oxygen functional group and water molecules into the crystal domains of GO. The diffractogram of GO also confirmed the presence of water molecules and oxygen functional groups into the GO layer [46]. After the thermal reduction of rGO at 100 °C, the interlayer spacing decreased from 0.826 to 0.805 nm, but the decrease was not significant. As the reduction temperature was increased from 100 to 200 °C, the interlayer spacing decreased significantly to 0.363 nm because rise in the reduction temperature increased the van der Waal’s forces existing between the stacked layers of rGO that eliminated the oxygen-containing functional groups from the layers of rGO crystal structure [47]. However, the interlayer spacing became almost constant with further increase in the reduction temperature from 200 to 600 °C.
FTIR analysis
The FTIR analysis was used to unravel the composition and functional groups present on the surface of Gr, GO and rGO and is shown in Fig. 2. The FTIR spectra of Gr showed peak at 2165 cm−1 and 2031 cm−1 attributed to the C≡C stretch of monosubstituted terminal alkyne and transition metal of carbonyl [48]. The spectra for GO were found to be similar to graphite. However, a broad peak at 3700–3000 cm−1 appeared in the GO spectra owing to the presence of hydroxyl groups and free radicals of water over the layer of GO [49]. The peak in the range of 1725–1700 cm−1 was assigned to the stretching vibration of C=O in carboxylic acid, carbonyl moieties and ketone present into the layer of GO. The appearance of peak at ~ 1620 cm−1 in the spectra of GO was due to the unoxidized graphitic domains, water impurity and scissor model assigned to the carbonyl, ketone and carbon double bond (C=C) [50]. A broad peak at 1550–1475 cm−1 was appeared in the GO due to the presence of asymmetric stretch of N–O compounds. The obtained peak near to 1039 cm−1 was attributed to stretching and bending vibration of carbonyls, hydroxyls, ethers, epoxides and ketones present between the stacked layers of GO structure.

Fourier transform infrared spectra of graphite, chemically reduced graphene oxide and thermally reduced graphene oxide at different temperatures
After the thermal reduction of GO at 100 °C, FTIR analysis of rGO-100 showed the disappearance of broadband appearing at around 3700–3000 cm−1 [51]. A new band at 1221 cm−1 was originated from a ketone, instead of epoxy which could be assigned to the symmetric and asymmetric stretching of S=O bonds in organic sulphates, i.e., tertiary alcohols, ester and sulphuric acid functional groups. It can be seen that the bands corresponding to C≡C (2165 cm−1), C=O (2001 cm−1) and C=C (1628 cm−1) were partially eliminated. The FTIR analysis of rGO obtained at higher reduction temperature, i.e., 300, 400 and 500 °C exhibited similar spectra. In the spectra of rGO-300, rGO-400 and rGO-500, the peaks conforming to C=O, N–O (1515 cm−1) and C–O (1039 cm−1) were completely removed due to the elimination of these functional groups from rGO at higher temperature. However, the disappearance of broadband occurring at 1628 cm−1 (C=O) was seen at 600 °C reduction temperature [48].
AFM analysis
The layer thickness, morphology and agglomeration of Gr, GO and rGO were evaluated from AFM analysis and further used to investigate the degree of exfoliation. Figure 3 shows the AFM images of graphite obtained before and after the chemical and thermal reduction process. AFM images revealed that the average thickness of samples was ~ 1 nm, which is a typical value for single layer of rGO sheet. Thickness of single layer of graphene sheet in suspended water had 0.334 nm, while exfoliated GO and rGO-100 both have ~ 0.80 nm layer thickness [10]. Such types of variation can be due to the presence of moisture, oxygen and carbonyl, epoxide, ketone-containing functional groups into the layer of graphite [52, 53].

AFM spectra of a graphite, b GO, c rGO-100, d rGO-200, e rGO-300, f rGO-400, g rGO-500 and h rGO-600
The XRD analysis of rGO also showed the exclusion of oxygen-containing functional groups from GO after the thermally reduction within the temperature range of 200–600 °C. The layer thickness of rGO in the temperature range of 200–500 °C was nearly 0.36 nm, while at 600 °C obtained layer thickness was 0.34 nm because at 500–600 °C most of the oxygen-containing functional groups were eliminated. AFM analysis also revealed that the rGO material contains few monolayers of graphite sheet in each particle after the thermal and chemical reduction.
Raman analysis
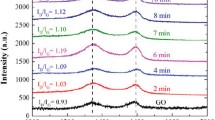
Raman analysis is widely used for the characterization of graphitic materials to find out the number of layers, ordered and disordered of the crystal structure, crystallite size of sp2 domain and various types of edge defects [54]. The spectra of GO and rGO illustrated the presence of G and D band peaks in the range of 1571–1585 cm−1 and 1346–1351 cm−1 which can be considered as an evidence for the presence of defects, i.e., order/disorder, vacancies, edge defects, etc. on carbon atom structure (Fig. 4). In pristine graphite, the intensity of G and D band peak were not visible in the range of 1571–1585 cm−1 and 1346–1351 cm−1 (Fig. 4a). In GO and rGO samples, it was observed that the D band narrowed, G band broadened and the variation in their peak positions along with the intensity showed the significance reduction of graphitic material [55]. However, after oxidation and thermal reduction of GO and rGO signified two most intense peaks, the first G band was in the range of at 1571–1585 cm−1 and second 2D or Gʹ band was in the range of 2685–2713 cm−1 (Fig. 4c). In the reduced graphene oxide G, Gʹ peaks are common and are allotted due to the E2g vibration mode of second order of the D band and sp2 domain of carbon atoms [56]. Tuinstra et al. [57] also reported the resonance of G band peaks corresponded to first-order scattering of the E2g vibration mode that exhibit Brillouin zone (K-point) centre in the optical mode of sp2 carbon domain (i.e., due to in plane vibration mode of carbon atoms). The second Gʹ band also known as 2D band can be originated from the double resonance Raman spectra and harmonic series of the D band. In this region, the single layer of rGO sheet was present in bilayer rGO that splits into tetrahedral form and reflects the evolution of the electron band structure of rGO. In the Raman spectra of Gr and rGO, 2D band peaks were active in the absence of defects [58]. In the GO and rGO, another peak was obtained in the range of 1346–1351 cm−1 attributed to D peak which could have been arisen from the first-order resonance. The A1g breathing mode of hexagonal carbon atoms near K-point was due to the presence of defects [24]. A defect activated peak known as D + G band also appeared at 2950 cm−1 due to the combination of different momenta with phonons near to full width half maximum (FWHM) and K-point. Defects are required for the activation of D band, which reveals the presence of impurities such as hydroxyl, epoxy, ester and carboxylic acid. In the initial graphite material, G and D band peaks were not visible in Raman spectra, while in GO the obtained peaks of G and D bands were at 1577, 1349 cm−1, which was related to order/disorder and structural defects of sp2 domains of carbon atoms [59, 60]. Upon oxidation and thermal reduction, the relative intensity of D and G band peaks, the width of 2D and D + G band peaks also decreased due to the breaking of AB stacking order in GO sample. Hong et al. also reported the increase in the relative intensity of D and G band peaks in rGO-100 and decrease in the width of 2D, D + G band as the reduction temperature was increased [61]. With an enhancement in the reduction temperature from rGO-200 °C to 600 °C, the relative intensity of D, G and 2D, D + G band peaks simultaneously decreased due to the influence of FWHM value. With the increment in the thermal reduction temperature, the significant variation in the FWHM value was due to the removal of defects from the rGO lattice structure [62]. With further increase in reduction temperature, the G band peak shifted towards a highest wave number, i.e., 1571–1584 cm−1. The shift in G band was due to the creation of sp3 carbon atoms over the GO and rGO structure because of the sp2 hybridization carbon atoms that behaved like a cluster in sp3 matrix [63].

Raman spectra of a graphite, b GO, c rGO-100, d rGO-200, e rGO-300, f rGO-400, g rGO-500 and h rGO-600
Various equations have been used to measure the average crystalline size (La), low defect density (LD) and defect density (ηD) of the sp2 domains of hexagonal carbon atoms. In this study, Tuinstra Koenig’s relation was employed to measure the average crystalline size. The low-density defects and defect density of the sp2 domains of carbon lattice structure were calculated from Knight and White relation [57, 64].
where λL was the wavelength of the Raman X-ray sources (532 nm) and ID/IG was the integrated intensity ratio between Raman D and G band peak areas. ID/IG ratio strongly depends on the excitation laser wavelength.
Table 2 shows that at lower reduction temperature the ID/IG ratio increased, which further decreased with the increase in reduction temperature. The increase in the ID/IG ratio shows plenty of structural defects over the rGO layer. The ID/IG ratio and calculated values across the plane from graphitic samples revealed the huge amount of sp3 domains is formed in rGO as compare to the GO.
XPS analysis
XPS analysis was used to examine the bonding interaction, hybridization of carbon atoms and chemical composition of Gr, GO and rGO samples. The core-level XPS spectrum of Gr, GO and rGO showed an intense peak at 284.1 eV and 530 eV, which corresponded to C1s and O1s elements (Fig. 5). The atomic concentration of C1s and O1s in Gr was found to be 96.31 and 3.69%, respectively [65, 66]. The absence of any other peaks in the core-level spectra of Gr excluded the presence of any other impurities in the prepared Gr.

Core-level XPS spectra of a graphite, b GO, c rGO-100, d rGO-200, e rGO-300, f rGO-400, g rGO-500 and h rGO-600
The core-level spectra of GO also displayed the peaks ascribing to C1s and O1s elements, but the atomic concentration of O1s in GO increased to 29.80% as compared to Gr (Fig. 5b). The increase in the atomic concentration was due to the introduction of oxygen-containing groups into the graphene backbone after the chemical oxidation. The C1s spectrum of GO presented well defines peaks centred at 284.27 eV and 286.35 eV attributing to the presence of C=C/C–C (aromatic rings of GO) and C–O functional groups (epoxy and hydroxyl groups), respectively (Fig. 6b) [67]. The deconvolution of C1s spectrum revealed three additional prominent peaks centred at 286.19, 287.81 and 290.11 eV assigned to the C–O–C (epoxy), C=O (carbonyl) functional groups and pi–pi* (π–π*) shake-up transitions groups [68].

C1s spectra of a graphite, b GO, c rGO-100, d rGO-200, e rGO-300, f rGO-400, g rGO-500 and h rGO-600
The core-level spectra of GO obtained after thermal reduction at 100 °C did not exhibited any significant change in the elemental composition of rGO-100 (Fig. 5c). The atomic concentration of C1s and O1s in rGO-100 was 70% and 30%, respectively. However, Paredes et al. [69] also reported the thermal reduction of GO at 200 °C; the atomic concentration of O1s in rGO-200 drastically reduced to 18.30% and the atomic concentration of C1s increased to 81.70%. This could be due to the reduction of GO that decreased the contents of oxygen-containing functional groups (C–O, C=O and COOH) in the rGO-200. Pei et al. also reported the C1s spectrum of rGO-300 also showed the characteristic peak of carbon at 284.51 eV [16]. The deconvolution of C1s revealed the fitting of five components peak at 284.46, 285.43, 286.44, 288.41, 289.50 and 289.74 eV (Fig. 6c). The peak at 284.46 eV corresponded to sp2 hybridization of carbon and sp3 hybridization of carbon in rGO-300 was seen at 285.44. The other peaks can be assigned to the epoxy C–O/hydroxyl C–OH (286.1–287.1 eV), carbonyl C=O (288.41 eV) and carboxylate O–C=O (289.5 eV), and low-intensity peak formed at 289.74 eV was due to pi–pi* (π–π*) shake-up transitions group [70, 71]. The interaction of oxygen-containing functional group with carbon was further confirmed from the deconvolution of O1s spectrum provided in Table 3. The fitting of O1s spectrum presented four main peaks centred at 530.0, 531.64, 532. 39 and 533.08 eV as shown in Fig. 7. The weak interaction of C=O with water in rGO-300 was observed at 530.0 and 531.64 eV, while the occurrence of hydroxyl and epoxy groups (C–OH/C–O–C) can be seen at 532.0 eV and 533.3 eV [72, 73]. The presence of theses peaks in the O1s spectrum may have been resulted from the incorporation of water into the stacked layers of carbon through the formation of hydrogen bonds via interaction of oxygen and/or hydrogen from water with carbon. The C1s spectrum of rGO obtained at 400, 500 and 600 °C reduction temperature exhibited approximately similar characteristics peaks for carbon. The fitting of C1s spectrum in rGO-400, rGO-500 and rGO-600 also displayed five prominent peaks at 284.46, 285.43, 286.44, 288.41, 289.50 and 289.74 eV corresponding to different carbon hybridizations (sp2 and sp3), bonding of oxygen groups to carbon (carboxyl, epoxy, carbonyl and carboxyl) and pi–pi* shake-up transitions group. The deconvolution of O1s spectrum also revealed four major peaks approximately centred at 529.0, 531.0, 532.0 and 533.0 eV conform to the presence of C=O (carbonyl, carboxyl) and C–O (hydroxyl, carboxyl, epoxy) functional groups [74].

O1s spectra of a graphite, b GO, c rGO-100, d rGO-200, e rGO-300, f rGO-400, g rGO-500 and h rGO-600
XPS analysis showed that GO and rGO-100 both have similar properties and high content of oxygen. As the reduction temperature was increased from 100 to 200 °C, a new broad peak corresponding to rGO appeared, due to the decrease in oxygen-based hydroxyl and epoxy groups from GO. With further increase in the reduction temperature to 300 °C, maximum amount of oxygen-containing functional groups was eliminated from GO. However, in the rGO-300 the atomic concentration of oxygen was found to be 19.60% which was due to the fraction amount of carboxyl functional groups left in the rGO. The rGO-400 showed 13.28% (atomic wt%) of oxygen because at 400 °C phenol groups were generated from the presence of ether groups in the GO. The rGO-500 and rGO-600 also exhibited the presence of oxygen content because carbonyls and phenols were generated in rGO-500 from the consumption of epoxy groups (C–O–C), and at 600 °C, the epoxy and hydroxyl groups were formed again over the rGO lattice structure.
Conclusions
GO was successfully synthesized from graphite powder using modified hummer method and thermal reduction of GO was achieved in tubular furnace under inert atmosphere by varying the reduction temperature. The FTIR, XRD, Raman and XPS analysis confirmed the presence of oxygen-containing functional groups, viz. hydroxyl, carbonyl, carboxyl and epoxy, in the lattice structure of GO. The thermal reduction process of GO eliminated these functional groups with a significant removal at 500 °C reduction temperature. The degree of elimination of oxygen-containing functional groups was found responsible for the decrease in the interlayer spacing of rGO, obtained at different reduction temperatures. FTIR analysis revealed the presence of hydroxyl groups, free radicals (water), unoxidized graphitic domains, water impurity and scissor models, which were assigned to the carbonyl, ketone, carbon double bond (C=C). The thermal reduction of GO at 200 °C showed disappearance of hydroxyl groups and partial elimination of C≡C, C=O and C=C functional groups, but the peaks conforming to C=O, N–O and C–O were completely removed at higher reduction temperature. Raman analysis of GO and rGO illustrated the presence of G and D band peaks in the range of 1571–1585 cm−1 and 1346–1351 cm−1 that disclosed the presence of defects, i.e., order/disorder, vacancies, edge defects on carbon atom structure. The core-level XPS spectra of Gr, GO and rGO revealed the characteristic peak of C1s and O1s elements and the absence of any other peaks confirmed the purity of prepared samples. The C1s spectrum of GO established the presence of C=C/C–C (aromatic rings of GO) and C–O functional groups (epoxy and hydroxyl groups), respectively, and the deconvolution of C1s spectrum revealed three additional prominent peaks assigned to the C–O–C (epoxy), C=O (carbonyl) functional groups and pi–pi* (π–π*) shake-up transitions groups. The C1s spectrum of rGO also exhibited similar characteristics peaks of carbon which confirmed the partial elimination of carbon bonded oxygen functional groups. The atomic concentration of carbon in rGO (~ 86%, w/w) was found higher as compared to chemically reduced GO owing to the disappearance of oxygen-containing functional groups from the stacked layer structure of GO.
References
A.K. Geim, K.S. Novoselov, Nat. Mater. 6, 183–191 (2007)
A.K. Geim, Science 324, 1530–1534 (2009)
K.S. Novoselov, A.K. Geim, S.V. Morozov, D. Jiang, Y. Zhang, S.V. Dubonos, I.V. Grigorieva, A.A. Firsov, Science 306, 666–669 (2004)
H.C. Lee, W.W. Liu, S.P. Chai, A.R. Mohamed, C.W. Lai, C.S. Khe, C.H. Voon, U. Hashim, N.M. Hidayah, Procedia Chem. 19, 916–921 (2016)
M. Khan, M.N. Tahir, S.F. Adil, H.U. Khan, M.R. Siddiqui, A.A. Al-warthan, W. Tremel, J. Mater. Chem. A 3, 18753–18808 (2015)
B.F. Machado, P. Serp, Catal. Sci. Technol. 2, 54–75 (2012)
C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A.N. Marchenkov, E.H. Conrad, Science 312, 1191–1196 (2006)
W. Qian, R. Hao, Y. Hou, Y. Tian, C. Shen, H. Gao, X. Liang, Nano Res. 2, 706–712 (2009)
M.J. Fernandez-Merino, L. Guardia, J.I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso, J.M. Tascon, J. Phys. Chem. C 114, 6426–6432 (2010)
C. Gomez-Navarro, R.T. Weitz, A.M. Bittner, M. Scolari, A. Mews, M. Burghard, K. Kern, Nano Lett. 7, 3499–3503 (2007)
S. Pei, J. Zhao, J. Du, W. Ren, H.M. Cheng, Carbon 48, 4466–4474 (2010)
S. Stankovich, D.A. Dikin, R.D. Piner, K.A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S.T. Nguyen, R.S. Ruoff, Carbon 45(7), 1558–1565 (2007)
D. Li, M.B. Muller, S. Gilje, R.B. Kaner, G.G. Wallace, Nat. Nanotechnol. 3(2), 101 (2008)
Y. Zhou, Q. Bao, L.A. Tang, Y. Zhong, K.P. Loh, Chem. Mater. 21, 2950–2956 (2009)
M. Veerapandian, M.H. Lee, K. Krishnamoorthy, K. Yun, Carbon 50, 4228–4238 (2012)
S. Pei, H.M. Cheng, Carbon 50, 3210–3228 (2012)
K.S. Novoselov, D. Jiang, F. Schedin, T.J. Booth, V.V. Khotkevich, S.V. Morozov, A.K. Geim, Proc. Natl. Acad. Sci. USA 102, 10451–10453 (2005)
Y. Si, E.T. Samulski, Nano Lett. 8, 1679–1682 (2008)
S. Stankovich, R.D. Piner, X. Chen, N. Wu, S.T. Nguyen, R.S. Ruoff, J. Mater. Chem. 16, 155–158 (2006)
D. Cai, M. Song, J. Mater. Chem. 17, 3678–3680 (2007)
J. Shen, Y. Hu, M. Shi, X. Lu, C. Qin, C. Li, M. Ye, Chem. Mater. 21, 3514–3520 (2009)
J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang, X. Zhang, Chem. Mater. 22, 2213–2218 (2010)
S. Stankovich, D.A. Dikin, G.H. Dommett, K.M. Kohlhaas, E.J. Zimney, E.A. Stach, R.D. Piner, S.T. Nguyen, R.S. Ruoff, Nature 442, 282–286 (2006)
D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R.D. Piner, S. Stankovich, I. Jung, D.A. Field, C.A. Ventrice, R.S. Ruoff, Carbon 47, 145–152 (2009)
H.J. Shin, K.K. Kim, A. Benayad, S.M. Yoon, H.K. Park, I.S. Jung, M.H. Jin, H.K. Jeong, J.M. Kim, J.Y. Choi, Y.H. Lee, Adv. Funct. Mater. 19, 1987–1992 (2009)
H.H. Huang, K.K.H. De Silva, G.R.A. Kumara, M. Yoshimura, Sci. Rep. 8, 6849 (2018)
C. Gomez-Navarro, J.C. Meyer, R.S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern, U. Kaiser, Nano Lett. 10, 1144–1148 (2010)
C. Mattevi, G. Eda, S. Agnoli, S. Miller, K.A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel, M. Chhowalla, Adv. Funct. Mater. 19, 2577–2583 (2009)
G.I. Titelman, V. Gelman, S. Bron, R.L. Khalfin, Y.B. Cohen, H. Bianco-Peled, Carbon 43, 641–649 (2005)
H. Wang, J.T. Robinson, G. Diankov, H. Dai, J. Am. Chem. Soc. 132, 3270–3271 (2010)
J.W. Suk, R.D. Piner, J. An, R.S. Ruoff, ACS Nano 4, 6557–6564 (2010)
B.C. Brodie, Philos. Trans. R. Soc. Lond. 149, 249–259 (1859)
T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis, I. Dekany, Chem. Mater. 18, 2740–2749 (2006)
B.S. Singu, K.R. Yoon, J. Alloys Compd. 770, 1189–1199 (2019)
N. Kumar, J.R. Rodriguez, V.G. Pol, A. Sen, Appl. Surf. Sci. 463, 132–140 (2019)
K.K. De Silva, H.H. Huang, R. Joshi, M. Yoshimura, Carbon 166, 74–90 (2020)
X. Gao, J. Jang, S. Nagase, J. Phys. Chem. C 114, 832–842 (2009)
W.S. Hummers, R.E. Offeman, J. Am. Chem. Soc. 80, 1339 (1958)
G. Venugopal, K. Krishnamoorthy, R. Mohan, S.J. Kim, Mater. Chem. Phys. 132, 29–33 (2012)
C. Hontoria-Lucas, A.J. Lopez-Peinado, J.D. Lopez-Gonzalez, M.L. Rojas-Cervantes, R.M. Martin-Aranda, Carbon 33, 1585–1592 (1995)
C.N. Rao, K. Biswas, K.S. Subrahmanyam, A. Govindaraj, J. Mater. Chem. 19, 2457–2469 (2009)
N.M. Hidayah, W.W. Liu, C.W. Lai, N.Z. Noriman, C.S. Khe, U. Hashim, H.C. Lee, AIP Conf. Proc. 1892, 150002 (2017)
S.K. Mishra, S.N. Tripathi, V. Choudhary, B.D. Gupta, Sens. Actuator B Chem. 199, 190–200 (2014)
F. Babak, H. Abolfazl, R. Alimorad, G. Parviz, Sci. World J. 2014, 10 (2014)
H.M. Ju, S.H. Choi, S.H. Huh, J. Korean Phys. Soc. 57, 1649–1652 (2010)
J. Chen, B. Yao, C. Li, G. Shi, Carbon 64, 225–229 (2013)
G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu, J. Yao, J. Phys. Chem. C 112, 8192–8195 (2008)
J. Coates, in Encyclopedia of Analytical Chemistry, ed. by R.A. Meyers, M.L. McKelvy (Wiley, Hoboken, 2006)
H. Fujimoto, Carbon 41, 1585–1592 (2003)
H. Wu, W. Zhao, H. Hu, G. Chen, J. Mater. Chem. 21, 8626–8632 (2011)
P.P. Jose, M.S. Kala, N. Kalarikkal, S. Thomas, Mater. Today Proc. 5, 16306–16312 (2018)
H.K. Jeong, Y.P. Lee, R.J. Lahaye, M.H. Park, K.H. An, I.J. Kim, C.W. Yang, C.Y. Park, R.S. Ruoff, Y.H. Lee, J. Am. Chem. Soc. 130, 1362–1366 (2008)
D. Lin, Y. Liu, Z. Liang, H.W. Lee, J. Sun, H. Wang, K. Yan, J. Xie, Y. Cui, Nat. Nanotechnol. 11, 626–632 (2016)
J. Kotakoski, A.V. Krasheninnikov, U. Kaiser, J.C. Meyer, Phys. Rev. Lett. 106, 105505 (2011)
C. Botas, P. Alvarez, C. Blanco, M.D. Gutierrez, P. Ares, R. Zamani, J. Arbiol, J.R. Morante, R. Menendez, RSC Adv. 2, 9643–9650 (2012)
S. Eigler, C. Dotzer, A. Hirsch, Carbon 50, 3666–3673 (2012)
F. Tuinstra, J.L. Koenig, J. Chem. Phys. 53, 1126–1130 (1970)
K.S. Vasu, B. Chakraborty, S. Sampath, A.K. Sood, Solid State Commun. 150, 1295–1298 (2010)
Y. Zhu, S. Murali, W. Cai, X. Li, J.W. Suk, J.R. Potts, R.S. Ruoff, Adv. Mater. 22, 3906–3924 (2010)
L.G. Cançado, A. Jorio, E.M. Ferreira, F. Stavale, C.A. Achete, R.B. Capaz, M.V. Moutinho, A. Lombardo, T.S. Kulmala, A.C. Ferrari, Nano Lett. 11, 3190–3196 (2011)
J. Hong, M.K. Park, E.J. Lee, D. Lee, D.S. Hwang, S. Ryu, Sci. Rep. 3, 2700 (2013)
A.C. Ferrari, J. Robertson, Phys. Rev. B 61, 14095 (2000)
J. Robertson, E.P. Oreilly, Phys. Rev. B. 35, 2946 (1987)
D.S. Knight, W.B. White, J. Mater. Res. 4, 385–393 (1989)
T.C. Chiang, F. Seitz, Ann. Phys. 10, 61–74 (2001)
F. Liu, H. He, Y. Ding, C. Zhang, Appl. Catal. B Environ. 93, 194–204 (2009)
M. Green, G. Marom, J. Li, J.K. Kim, Macromol. Rapid Commun. 29, 1254–1258 (2008)
Y. Shao, J. Wang, M. Engelhard, C. Wang, Y. Lin, J. Mater. Chem. 20, 743–748 (2010)
J.I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso, J.M. Tascon, Langmuir 25, 5957–5968 (2009)
J. Cao, G.Q. Qi, K. Ke, Y. Luo, W. Yang, B.H. Xie, M.B. Yang, J. Mater. Sci. 47, 5097–5105 (2012)
S. Park, R.S. Ruoff, Nat. Nanotechnol. 4, 217–224 (2009)
L.Z. Fan, J.L. Liu, R. Ud-Din, X. Yan, X. Qu, Carbon 50, 3724–3730 (2012)
S. Kundu, Y. Wang, W. Xia, M. Muhler, J. Phys. Chem. C 112, 16869–16878 (2008)
B.V. Crist, XPS Reports 1 (2007)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare that we have no known competing financial interests or personal relationships that could appear to influence the work reported in this manuscript.
Rights and permissions
About this article

Cite this article
Kumar, P., Divya, N. & Ratan, J.K. Study on the physico-chemical properties of reduced graphene oxide with different degrees of reduction temperature. J IRAN CHEM SOC 18, 201–211 (2021). https://doi.org/10.1007/s13738-020-02014-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-020-02014-w