
Abstract
In the present work, a novel platform has been designed to fabricate an electrochemical sensor composed of chitosan nanofibers modified with sodium dodecyl sulfate, a negatively charged surfactant, and hemoglobin protein for direct and sensitive determination of hydrogen peroxide. Chitosan nanofibers were synthesized through the electrospinning process and characterized by Fourier transform infrared spectroscopy and scanning electron microscopy. The synthesized nanofibers provided a large surface for the adsorption of hemoglobin. Immobilization of hemoglobin on the fibers was investigated by cyclic voltammetry, and the modification steps were followed by electrochemical impedance spectroscopy. Under the optimal conditions, the fabricated biosensor was employed for electrocatalytic determination of hydrogen peroxide by differential pulse voltammetric and amperometric methods in the linear range 3.0–2940.0 µM (detection limit = 0.16 µM) and 1.0–201.0 µM (detection limit = 0.05 µM), respectively. The applicability of the constructed biosensor was also evaluated by hydrogen peroxide determination in real samples, and satisfactory recoveries were obtained which confirmed that the proposed method provides a simple and sensitive method for the hydrogen peroxide monitoring.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrogen peroxide (H2O2) is a very important species for pharmaceutical, textile and food industries as well as clinical, environmental and mining applications [1]. H2O2 is very stable reactive oxygen species which can be found in biochemical systems due to the oxygen reduction. It is also formed as a byproduct of oxygen reduction reaction in cell processes, in the presence of enzymes such as catalase which use the H2O2 ability for the oxidation of desired substrates [2]. It can also induce oxidative damage to many cellular components if it exists in excess under stress conditions. Additionally, ·OH as the most potent and reactive oxygen species (ROS), which causes the oxidative degradation of biological macromolecules such as lipids, proteins, carbohydrates and nucleic acids, can be generated through the conversion of H2O2 [3]. Therefore, development of H2O2 analysis methods has great importance [1]. Many techniques including spectrofluorimetry [3], chemiluminescence [4], colorimetric [5], surface-enhanced Raman spectroscopic [6] and electrochemical methods [7, 8] have been reported for the determination of H2O2 [9]. Among these techniques, electrochemical methods owing to their selectivity, sensitivity, rapidity and simplicity have attracted much attentions [8,9,10]. Electrochemical sensors for H2O2 detection are categorized into two types of enzymatic, which mainly uses a peroxidase as the enzyme, and non-enzymatic, which is based on direct electroreduction in H2O2 [11]. However, enzymatic sensors suffer from several limitations, including complex procedures, narrow response ranges, high costs of fabrication, as well as their limited lifetime and stability. Therefore, development the non-enzymatic sensor for the sensitive and selective determination of H2O2 in a wide concentration range by straightforward process is necessary [9]. Although H2O2 can participate in direct redox processes at the electrode surface, these processes cannot be applied for analytical purposes due to its slow electron transfer and high over-potentials at the electrode surface. Applying the high voltage causes serious interferences from other electroactive species in real matrices. The improvement in the conductivity and the catalytic performance is still required to enhance the properties to satisfy the realistic applications of the non-enzymatic sensors.
Heme proteins such as hemoglobin (Hb), due to their redox capability, have the great potential for using in the electrochemical biosensors. These proteins are a category of metalloproteins containing iron-centered porphyrin and can catalyze H2O2-dependent oxidation reactions [12,13,14,15]. As Hb is commercially available, its structure is known and its cost is moderate, it is widely used as a model in the study of the direct electron transfer of proteins [12].
Nanomaterials are used for modification of the electrodes for the study of the direct electrochemistry of the redox proteins and also to construct third generation biosensors [7]. Nanofibers (NFs) with their unique properties including very large specific surface area and high porosity with very small pore size are the very important class of nanomaterials which are widely used in sensor applications [13]. Electrospinning is a versatile and efficient method for the fabrication of various NFs [14]. When diameters of polymeric fiber shrink from micrometer range to nanometer ranges, several amazing characteristics appear [18].
Chitosan (Cs), a natural cationic polysaccharide, is still an interesting polymer due to its physicochemical properties, including biodegradability, biocompatibility and non-toxicity. It is used as a safe material for use in biomedical applications [15, 16].
In this work, we reported on a low cost and straightforward method for fabrication of a non-enzymatic biosensor for H2O2 detection through protein immobilization on a novel and effective platform. An anionic surfactant (sodium dodecyl sulfate, SDS) was utilized to modify the surface of the Cs nanofiber (CsNF) for Hb entrapment. This strategy leads to efficient protein stabilization by facilitating the accessibility to the protein redox group. Finally, a gold electrode surface was modified by CsNF-SDS-Hb nanostructure and used for electrochemical detection of H2O2.
Experimental
Reagents and apparatus
Electrochemical measurements were conducted in phosphate buffer solutions using a Polarography device (Metrohm model 797) and Autolab 302 N potentiostat/galvanostat (Metrohm Autolab B.V., The Netherlands). A conventional three-electrodes setup including a platinum wire as the counter electrode, an Ag/AgCl as the reference electrode and the modified gold electrode as the working was used for electrochemical measurements. The FTIR spectrum was recorded by a Perkin-Elmer spectrophotometer (model Spectrum GX). The morphology and structure of the synthesized materials were characterized by scanning electron microscopy (SEM, Philips, XL30, Netherland). The electrospinning experiments were carried out with an Electrospinning machine (Model Full option lab2 ESI-II, Fanavaran Equipment Nanoazma Co., Iran). The pH values were carefully adjusted by addition of diluted HCl or NaOH solutions and measured with a pH-meter (Metrohm model 827).
Cs (degree of deacetylation 0.85%, MW 110 kDa), Hb, SDS, hydroxybenzotriazole monohydrate (HOBt.H2O), polyvinyl alcohol (PVA) (degree of polymerization = 1600, degree of hydrolysis = 97.5–99.5 mol%) were purchased from Sigma-Aldrich and Fluka. All other materials were of analytical grade and were used without further purification. Double-distilled water was used for the aqueous solutions preparation, and the stock solutions of H2O2 were freshly prepared.
Preparation of electrospinning stock solutions
A 2% (w/v) Cs solution was prepared by dissolving Cs, and HOBt at a weight ratio of 1:1 in distilled water (0.1 g of HOBt was added to 90 mM of Cs solution under stirring for 4 h). The 10% (w/v) PVA solution was prepared by dissolving PVA in distilled water at 80 °C, and then the solution was stirred with a magnetic stirrer for 4 h. The 2% Cs-HOBt solution was mixed with a 10% PVA solution at weight ratios of 70/30 [15].
Electrospinning process
The electrospinning system used in this study contained Hamilton syringe with stainless steel needle (diameter of 0.9 mm) which was connected to the positive polarity emitting electrode of high-voltage DC power supply (20–30 kV). A wrapped aluminum sheet on a rotating collector with a diameter of 0.5 mm, which was rotating with controllable speed, was used for the collection of fibers. The distance between the tip of the syringe and collector was 12 cm, and the applied potential was fixed at 15 kV. With taking the prepared solution of polymer in the syringe and injecting it in the capillary needle, the jet initiation was achieved: By charging the solution followed by the injection, the polymer solution was ejected and drowned toward the collator as a whipping jet. The solution feed of the polymer was driven by the electrostatic forces generated at the spinning. Meanwhile, with the evaporation of the solvent, continues polymer fiber accumulated on the aluminum foil. All spinning process was conducted at room temperature.
Preparation of CsNF-SDS-Hb
Basically, surface modification of the electrodes can be accomplished by a coating of the surface through chemical or physical adsorption of the desired molecules, depending on the specific applications [7]. To form the CsNF-SDS-Hb suspension, SDS, CsNF and Hb with desired concentration ratios ([Hb]/[SDS] = 0.2, [CsNF]/[SDS] = 0.003). In details, 3 mg of CsNFs was dispersed in diluted HCl 1.0 mM for 30 min. Then, SDS reagent (0.3 M) was prepared and mixed with it. Finally, the Hb solution was added to the mixture and mixed for 1 h.
Fabrication of the proposed biosensor
First, in order to obtain a mirror-like surface, the gold electrode (diameter = 3 mm) was polished with 0.3 and 0.05 μm alumina and then ultrasonicated for 5 min in deionized water and ethanol solutions, respectively, and then it was allowed to dry at room temperature [17]. Afterward, 10 μL of the prepared CsNFs-SDS-Hb was dropped on the cleaned surface of the gold electrode and was dried at room temperature for 2 h. The CsNFs-SDS-Hb modifier is attached to the electrode surface via the self-assembling of amino groups of Cs on the Au electrode surface. Then, for removing unbounded nanostructures, the surface of the Au electrode was rinsed thoroughly with deionized water. Finally, the fabricated biosensor was stored at refrigerator (4 °C, in a phosphate buffer solution of pH 7.4) before use. Schematic representation of the fabrication process of the proposed biosensor is shown in Scheme 1.

The fabrication process of the proposed biosensor
Preparation of real sample
Since H2O2 is often used as a preservative agent in milk, for the real sample analysis, the applicability of the proposed method was examined for the detection of H2O2 in milk samples [18]. A sufficient sterilization of milk is considered at 0.03% H2O2 content [19]. By standard addition method, H2O2 content in milk sample was determined using the proposed biosensor [18,19,20].
A 0.5 mL of a fresh milk sample was added to 4.5 mL of phosphate buffer solution. The milk samples were spiked with various concentrations of H2O2, and then non-spiked and spiked samples were analyzed. In order to confirm the accuracy of the method, the control experiment was used by the classical potassium permanganate titration as the standard spectrophotometric method [18, 20].
Results and discussion
Characterization of synthetic nanofibers by FTIR and SEM
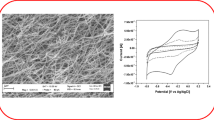
To realize the successful synthesis of NFs, FTIR was used to investigate the Cs-HOBt/PVA nanofibers (CsNFs) blended in a weight ratio of 30/70 (Cs/HoBt: PVA). The FTIR spectrum of CsNFs (Fig. 1a) exhibits absorption peaks at 1538, 1630 and 1680 cm−1 related to ammonium ion, scissor vibration of the amine and C=O stretching of the N-acetyl groups, respectively [21].

a FTIR and b SEM image of CsNFs
Surface morphology and geometrical properties of NFs such as fiber diameter and diameter distribution were characterized using SEM, and results are shown in Fig. 1b. The SEM micrograph confirms the formation of a uniform morphology virtually bead-free nanofibers, electrospun from the polymer solution. As can be seen in the figure, the range of nanofiber diameters is between 200 and 500 nm. Among them, some of the nanofibers show a narrow distribution around 200 nm diameters that increased the surface area. Also, there is a lot of the available spaces between the nanofibers that caused the fast diffusion of analyte to the surface.
Characterization of the fabricated biosensor
Another important issue for the development of a biosensing platform is the preparation process of the biosensor [17]. Electrochemical impedance spectroscopy (EIS) is a non-destructive and real-time measurement method for analyzing the interfacial properties of the surface of modified electrodes in the sensor fabrication processes [22]. Therefore, changes in the charge transfer resistance (RCT) after each modification step were monitored by EIS. RCT controls the electron transfer kinetics of the redox probe at the electrode interface. Its value varies when different substances are adsorbed onto the electrode surface [22].
The illustrated Nyquist plots in Fig. 2a obtained from different modification steps of the sensing layer. For accurate estimation of RCT at the electrode surface, an equivalent circuit (Randles’ equivalent circuit) was used for fitting of EIS and the obtained results from the Nyquist plot [22]. The fitting of EIS data and experimental data was shown in the figure by a solid line and dot plots, respectively. As shown in the figure, the estimated RCT at the bare Au electrode was evaluated about 417 Ω that after modification by CsNFs-SDS-Hb was increased to 1280 Ω. Also, the obtained Nyquist plots from modified AuE by CsNFs and Hb were shown in figure. As can be seen in the figure, surface electrode modification with CsNFs leads to the increase in RCT (887 Ω) while adding the Hb at the bare AuE does not lead to the meaningful changing in RCT (489 Ω). It means that Hb cannot be effectively addition to the AuE surface without the presence of CsNFs.

a Nyquist plots of different fabrication steps of biosensor (in PBS containing 5 mM K3[Fe(CN)6]/K4[Fe(CN)6] (1:1) and 0.1 M KCl), b CVs of Au electrodes (AuE) modified with CsNFs, CsNFs-Hb and CsNFs-SDS-Hb in a 0.1 M PBS (pH 7.0) containing 0.1 M KNO3 at a scan rate of 100 mV s−1, and c DPVs of Bare AuE in PBS containing 2940 μM of H2O2, CsNFs-SDS-Hb/AuE in PBS and CsNFs-SDS-Hb/AuE in PBS containing the 2940 μM of H2O2 (the voltammograms were obtained at pH 7)
Direct electrochemistry of CsNFs-SDS-Hb/Au electrode
Hb immobilization on the CsNFs was investigated by recording the cyclic voltammograms (CV) of bare AuE, CsNFs/AuE, CsNFs-Hb/AuE and CsNFs-SDS-Hb/AuE in 0.1 M phosphate buffer solution containing 0.1 M KNO3, as the supporting electrolyte, at a scan rate of 100 mV s−1. As seen in Fig. 2b, bare AuE, CsNFs/AuE and CsNFs-Hb/AuE show no electrochemical signal. It means that no redox reaction took place at the electrode surface. There were not any Hb peaks observed in the absence of SDS. At the CsNFs-SDS-Hb/AuE, a reduction peak was observed, which is an indication of Hb immobilization [12]. Comparing the voltammograms for CsNFs-SDS-Hb/AuE with CsNFs-Hb/AuE, the reduction peak which is appeared for CsNFs-SDS-Hb/AuE indicates the main contribution of SDS in the Hb immobilization and therefore in the enhancing of Hb direct electron transfer. Since the surface of the synthesized chitosan NFs is positively charged, the anionic SDS surfactants can be electrostatically adsorbed at its surface. The protein isoelectric point (pI) is given by the pH value at which the zeta potential is approximately zero [23]. At the pH values below the Hb pI (was reported around 5.5 [23]), there are cationic charges on it and thus Hb with its net positive charge adsorbs strongly to the negatively charged membrane[23,24,25].
Voltammetric behavior of H2O2 at CsNFs-SDS-Hb/AuE
Hb due to its close similarity to peroxidase has certain intrinsic peroxidase activity and therefore, can be employed to catalyze the H2O2 reduction reaction [12]. To investigate the H2O2 reduction at the modified electrode surface, DPV method was used and recorded voltammograms are shown in Fig. 2c. No redox peaks were observed on the CsNFs-SDS-Hb/AuE in the absence of H2O2. At the bare AuE and the CsNFs-SDS-Hb/AuE, in the presence of 2940 μM of H2O2, a reduction peak was observed, which is assigned to the peroxide reduction. As can be seen in the figure, at the CsNFs-SDS-Hb/AuE surface, the electroreduction peak potential is shifted to more negative potentials and the peak current is increased. Obtained results demonstrated that CsNFs-SDS-Hb could display great electrocatalytic activity to peroxide.
Optimization of the condition
Optimization of the Hb immobilization condition
Response current is dependent not only on the concentration of H2O2 but also on the concentration of immobilized Hb in the film. Therefore, the ratios between Cs, SDS and Hb in the film are very important for optimal sensor operation [26]. Different amounts of CsNFs (1, 2, 3, 4, 5 and 6 mg mL−1, Fig. 3a), SDS (50, 100, 200, 260 and 325 mg mL−1, Fig. 3b), and Hb (10, 15, 20, 25, 30 and 40 mg mL−1, Fig. 3c) were used in 1 mL solutions that were applied for open circuit attachment of Hb at the nanofiber surface. To form the CsNFs-SDS-Hb suspension, CsNFs, SDS and Hb were mixed using an ultrasonic bath and the reduction current of CsNFs-SDS-Hb on the Au electrode surface was monitored by CV measurements. Through the obtained results from experiments, the optimal concentration ratio between SDS and Hb, at the amount of 3 mg mL−1 of CsNF, was found the [SDS]/[Hb] = 5.

Optimization of the amount of a CsNFs, b SDS and c Hb to form the CsNFs-SDS-Hb suspension, d Differential pulse voltammograms of the biosensor toward 600 µM of H2O2 at different pHs (inset shows the related changes of DPV response at different pHs)
Optimization of pH
Since the solution pH has a strong effect on the electrochemical activity of Hb, its effect on the response of Hb-based sensors is also remarkable [27]. To find the suitable pH for biosensor operation, pH of the solution was investigated from 5.0 to 9.0 in phosphate buffer solution and DPV responses of the biosensor toward 600 µM of H2O2 were recorded at different pHs (Fig. 3d). This figure shows that with increasing pH to 7.0, peak current (Ip) increases and then decreases, and the maximum peak current was achieved at pH 7.0. The biosensor shows lower catalysis ability in acidic environments than in alkaline environments [26]. Therefore, pH 7.0 was chosen as the optimum pH value and further experiments were conducted in this pH [26].
Analytical performance of the proposed biosensor
Calibration curve
Under the optimized experimental conditions, DPVs of the biosensor for various H2O2 concentrations were recorded and are shown in Fig. 4a. Quantitative analysis was based on the linear correlation between the concentration of H2O2 and Ip. The obtained calibration curve for H2O2, as shown in Fig. 4b contains two linear segments: the first linear part increases from 3.0 to 250 µM with the regression equation of Ip (µA) = 0.131 Chydrogen peroxide (µM) + 0.7722 (R2 = 0.9849), and the second linear part increases up to 2940 µM with the regression equation of Ip (µA) = 0.0436 Chydrogen peroxide (µM) + 23.368 (R2 = 0.9989). The detection limit of the proposed biosensor, based on 3Sb/m, was estimated to be 0.16 µM.

a and b obtained DPVs of CsNFs-SDS-Hb/AuE for various H2O2 concentrations under optimum conditions and the corresponding calibration curve, c, and d obtained an amperometric response and related calibration curve for 10 μM addition of H2O2 for each step at the CsNFs-SDS-Hb/AuE surface
Also, the amperometric response of the proposed biosensor toward the various concentration of H2O2 was recorded (Fig. 4c). The CsNFs-SDS-Hb/AuE offered an excellent, quick and high sensitive linear amperometric response toward the hydrogen peroxide in the 1˗201 μM concentration range with a detection limit of 0.05 μM (Fig. 4d).
A comparison between the present method and other reported electrochemical methods for the H2O2 determination is provided in Table 1. This comparison reveals that the obtained figures of merit for a proposed biosensor are improved in comparison with the many reported methods.
Repeatability, reproducibility and stability
The repeatability of the developed electrode was investigated by five repetitive measurements of 120 and 1780 µM H2O2 and 2.1% and 1.4% for RSD % were obtained, respectively.
Reproducibility of the proposed biosensor was investigated at H2O2 concentration of 1500 µM. RSD % for five biosensors, which were constructed at the same conditions obtained 2.8%.
Furthermore, the stability properties of the presented biosensor were evaluated. After a period of five weeks, 95% of the original response of the electrode to 1190 µM H2O2 remained.
These obtained results indicated that the prepared biosensor has suitable repeatability, reproducibility and stability for H2O2 determination.
Interference studies
In order to investigate the selectivity of the proposed method toward H2O2, interference effect of some cations and anions which might be found in biological liquids was investigated under the optimized conditions. Various concentrations of each species were added to the 150 µM H2O2 solution. The tolerance limit was taken as the maximum concentration of foreign species, which causes a relative error of less than approximately 5% [28]. The given results in Table 2 confirm that most of the investigated compounds are not affecting the H2O2 determination, and proposed biosensor can perform in the real samples with no significant interference matrix effect.
Real sample analysis
Since H2O2 is often used as a preservative in milk, milk samples were chosen in order to demonstrate the applicability of the proposed biosensor in real samples. The standard addition method was applied for the determination of H2O2 concentrations in milk samples [18, 20]. Four different milk samples were collected for this assay and spiked with 2 µM of H2O2. The results are listed in Table 3.
Moreover, using a classical potassium permanganate titration as the most exact and reliable analytical procedure for H2O2, the accuracy of the proposed method was confirmed [20]. Obtained results in Table 3 prove that the proposed biosensor can be efficiently used for the H2O2 determination in real samples.
Conclusions
In this work, an electrochemical biosensor based on Au electrode modified with CsNFs-SDS-Hb was fabricated and used for direct, sensitive and selective determination of H2O2. The obtained results demonstrated that electrode modification with NFs provides a suitable matrix for Hb immobilization on the electrode surface, and therefore, the proposed biosensor showed a good performance in H2O2 bioelectrocatalytic reduction. Also, the proposed biosensor exhibited a low limit of detection, with a wide linear concentration range, along with suitable repeatability, reproducibility and storage stability.
References
W. Chen, S. Cai, Q.-Q. Ren, W. Wen, Y.-D. Zhao, Analyst 137, 49 (2012)
N. Wang, C.J. Miller, P. Wang, T.D. Waite, Anal. Chim. Acta 963, 61 (2017)
W. Cui, H. Qin, Y. Zhou, J. Du, Microchim. Acta 184, 1103 (2017)
Y.B. Tsaplev, J. Anal. Chem. 67, 506 (2012)
S. Ge, W. Liu, H. Liu, F. Liu, J. Yu, M. Yan, J. Huang, Biosens. Bioelectron. 71, 456 (2015)
L.-L. Qu, Y.-Y. Liu, S.-H. He, J.-Q. Chen, Y. Liang, H.-T. Li, Biosens. Bioelectron. 77, 292 (2016)
H. Bagheri, E. Ranjbari, M. Amiri-Aref, A. Hajian, Y.H. Ardakani, S. Amidi, Biosens. Bioelectron. 85, 814 (2016)
Y. Zhang, B. Huang, F. Yu, Q. Yuan, M. Gu, J. Ji, Y. Zhang, Y. Li, Microchim. Acta 185, 86 (2018)
P. Arul, S.A. John, Electrochim. Acta 235, 680 (2017)
H. Dai, W. Lü, X. Zuo, Q. Zhu, C. Pan, X. Niu, J. Liu, H. Chen, X. Chen, Biosens. Bioelectron. 95, 131 (2017)
V. Muhr, M. Buchner, T. Hirsch, D.J. Jovanović, S.D. Dolić, M.D. Dramićanin, O.S. Wolfbeis, Sens. Actuators B 241, 349 (2017)
H. Fan, S. Zhang, P. Ju, H. Su, S. Ai, Electrochim. Acta 64, 171 (2012)
X. Geng, O.-H. Kwon, J. Jang, Biomaterials 26, 5427 (2005)
D. Li, Y. Xia, Nano Lett. 4, 933 (2004)
N. Charernsriwilaiwat, P. Opanasopit, T. Rojanarata, T. Ngawhirunpat, P. Supaphol, Carbohydr. Polym. 81, 675 (2010)
K. Ohkawa, K.-I. Minato, G. Kumagai, S. Hayashi, H. Yamamoto, Biomacromol. 7, 3291 (2006)
A. Afkhami, P. Hashemi, H. Bagheri, J. Salimian, A. Ahmadi, T. Madrakian, Biosens. Bioelectron. 93, 124 (2017)
B. Wang, J. Zhang, G. Cheng, S. Dong, Anal. Chim. Acta 407, 111 (2000)
D.S. Jeykumari, S. Ramaprabhu, S.S. Narayanan, Carbon 45, 1340 (2007)
D. Ye, H. Li, G. Liang, J. Luo, X. Zhang, S. Zhang, H. Chen, J. Kong, Electrochim. Acta 109, 195 (2013)
S. Haider, S.-Y. Park, J. Membr. Sci. 328, 90 (2009)
P. Hashemi, A. Afkhami, H. Bagheri, S. Amidi, T. Madrakian, Anal. Chim. Acta 984, 185 (2017)
P.S. Santiago, F.A.O. Carvalho, M.M. Domingues, J.W.P. Carvalho, N.C. Santos, M. Tabak, Langmuir 26, 9794 (2010)
B. Goldberg, A. Stern, Arch. Biochem. Biophys. 178, 218 (1977)
S. Onder, L.M. Schopfer, J.R. Cashman, O. Tacal, R.C. Johnson, T.A. Blake, O. Lockridge, Anal. Chem. 90, 974 (2017)
S. Yao, J. Xu, Y. Wang, X. Chen, Y. Xu, S. Hu, Anal. Chim. Acta 557, 78 (2006)
J. Zhang, M. Oyama, Electrochim. Acta 50, 85 (2004)
P. Hashemi, H. Bagheri, A. Afkhami, S. Amidi, T. Madrakian, Talanta 176, 350 (2018)
P.N. Mashazi, K.I. Ozoemena, T. Nyokong, Electrochim. Acta 52, 177 (2006)
A. Kafi, G. Wu, A. Chen, Biosens. Bioelectron. 24, 566 (2008)
F. Wang, R. Yuan, Y. Chai, Eur. Food Res. Technol. 225, 95 (2007)
Z. Tong, R. Yuan, Y. Chai, Y. Xie, S. Chen, J. Biotechnol. 128, 567 (2007)
B. Wang, J.-J. Zhang, Z.-Y. Pan, X.-Q. Tao, H.-S. Wang, Biosens. Bioelectron. 24, 1141 (2009)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kholosi, F., Afkhami, A., Hashemi, P. et al. Bioelectrocatalysis and direct determination of H2O2 using the high-performance platform: chitosan nanofibers modified with SDS and hemoglobin. J IRAN CHEM SOC 17, 1401–1409 (2020). https://doi.org/10.1007/s13738-020-01865-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-020-01865-7