
Abstract
A new electrochemical biosensor is developed for the detection of diazinon. For this purpose, a glassy carbon electrode is modified with MWCNTs and poly-l-lysine to immobilize a double-strain DNA (ds-DNA) on the surface of the electrode. In the first step, the interaction of diazinon with ds-DNA is transduced by electrochemical impedance spectroscopy and UV–Vis spectroscopy to monitor the intercalation of diazinon with DNA helix. This interaction leads to reduced interfacial charge-transfer resistance (Rct). The difference in the Rct before and after the interaction is considered as a suitable signal for diazinon detection. The proposed biosensor has a low detection limit (0.3 nmol L−1), a wide linear dynamic range (0.001‒100 µmol L−1), and high selectivity for the determination of diazinon. Finally, the performance of the biosensor for detecting of diazinon is verified in real samples such as river water, agricultural wastewater, lettuce juice, and tomato juice.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Organophosphate (OP) compounds have been widely studied for their toxicity, persistence, and storage [1]. A group of phosphoric acids with ester derivatives in their structures are OP compounds that are most commonly used as insecticides. According to researches, all the structure of the OP insecticides (P=S and P=O) metabolites have genotoxic properties (Scheme 1). Diazinon (o,o-diethyl o-(2-isopropyl-4-methyl-6-pyrimidyl) phosphorothioate) is a nonsystemic organophosphorus insecticide that finds its way into a variety of agricultural products such as lettuce, almond, citrus, cotton, leather, and alfalfa [2]. It has been used as a pesticide in agriculture. Diazinon and its derivatives are known as toxic materials in nature [3]. Hence, their quantitative and qualitative determination is important for environmental protection. Diazinon breaks down into an active oxon-metabolite in the environment or in living organisms to form diazoxon (o,o-diethyl o-(2-isopropyl-4-methyl-6-pyrimidinyl) phosphate), which inactivates acetylcholinesterase (AChE) as a responsive enzyme that disables neurotransmitter acetylcholine [4]. Inactivation of AChE causes the neurotransmitter acetylcholine to accumulate in the spaces between cholinergic synapses, leading to synaptic obstruction and reduced signal transmission [5].

Molecular structures of organophosphate compounds with a group of phosphoric acids with ester derivatives
A variety of methods such as high-performance liquid chromatography [6], immunoassay [7], spectrophotometry [8], infrared spectroscopy [9], enzymatic techniques [10], and gas chromatography [11] have been used for measuring diazinon. These methods are, however, mostly time-consuming and labour-intensive while they also require expensive equipment. In contrast, electrochemical methods are superior due to their sensitivity, simplicity, and low cost [12]. One such method that has recently attracted a lot of attention is the determination of analytes using modified electrodes in which nucleic acids are used as one of the best tools in the detection [13]. Study of interactions between ds-DNA and molecules or ions provides useful information. These interactions cause changes in the structure of ds-DNA in three different ways: (1) electrostatically, which involves a nonspecial interaction through an external linkage with the ds-DNA helix, (2) groove binding, in which the compounds are directly bonded to the edges of the base pairs in the primary and secondary grooves of ds-DNA, and (3) intercalation, in which flat combinations are placed between the base pairs and cause the base pair to unwind [14].
An important factor in the fabrication of DNA electrochemical biosensors is the immobilization of ds-DNA at different sites on the electrode surface to enhance the biosensor’s stability, reproducibility, and sensitivity. In 1991, carbon nanotubes (CNTs) with interesting and weird features were discovered that led to significant developments in various fields of science, especially in nanotechnology [15]. One of their important features is the high specific surface area, which enhances electron transfer. Thus, when used in electrochemical methods, they increase electrode sensitivity. Nevertheless, immobilizing of ds-DNA on CNTs is not appropriate because the negatively charged functionalized CNTs repel the negative charge of ds-DNA. A simple way to resolve this problem is using polymer films (with a neutral or positive charge) at the electrode surface, before immobilizing of the DNA.
In this study, electrostatic binding of the phosphate groups of the ds-DNA to poly-l-lysine (PLL), with amino groups, is exploited to modify the electrode [16, 17]. In this way, the surface of a glassy carbon electrode (GCE) is initially modified with carboxylic group-functionalized multiwall carbon nanotubes (MWCNTs) to form MWCNT/GCE. PLL films are then electro-polymerized at the surface of MWCNTs/GCE through cyclic voltammetry (CV) to form PLL/MWCNTs/GCE. Here, negative charge of the phosphate groups of the ds-DNA easily binds with the positive charge of PLL with a strong electrostatic interaction. Scanning electron microscope (SEM), UV–Vis spectroscopy, electrochemical impedance spectroscopy (EIS), and cyclic voltammetry are used to characterize the immobilized ds-DNA at the electrode surface. EIS was used as a suitable tool to detect diazinon by using the difference in the charge-transfer resistance of the biosensor before and after the interaction with diazinon.
Experimental
Material and reagents
All reagent-grade chemicals such as salmon sperm ds-DNA, Tris–HCl, CH3COOH, CH3COONa, EDTA, NaCl, H3PO4, DMF, K3Fe(CN)6, K4Fe(CN)6, (S)-2,6-diaminocaproic (l-lysine), ethanol, NaOH, Na2SO4, CaCO3, Pb(NO3)2, KIO3, Al(NO3)3, Cu(NO3)2, NiCl2.6H2O and Mn(CH3COO)2 were purchased from Aldrich (Germany). MWCNTs (with diameters in the range of 70 and 110 nm) and all of the other organic compounds were purchased from Fluka. The G–C% content of the DNA was reported to be 41.2%. The stock solution of ds-DNA (1000 mg L−1) in Tris buffer (pH 7.0) was prepared and kept frozen before the acetate buffer (pH 4.8) was used for its further dilution. The diazinon solution was prepared and maintained at 4 °C. Diluted diazinon solution was prepared by Tris–HCl buffer solution (pH 7.0). Stock solutions of amitrole (3-amino-1,2,4-triazole), monolinuron, carbaryl, monilate, chlorpyrifos (1000 mg L−1) were prepared by dissolving accurately weighed amounts of these compounds in ethanol. Fenitrothion and dichlorvos stock solutions (1000 mg L−1) were prepared by diluting the original pesticide solutions with methanol.
Apparatus
Electrochemical measurements were taken using Bio-logic SAS (SP-300) with EC-Lab (version 10.38). A three-electrode cell contains Ag/AgCl (3.0 mol L−1 KCl) as a reference electrode, a platinum wire as a counter electrode, and a modified glassy carbon electrode (GCE), with a cross section of 0.0314 cm2, as a working electrode. UV–Vis spectrometry was performed on a double-beam spectrophotometer, Jasco Model V-750, using 1.0-cm quartz cells. Scanning electron microscopic (SEM) images were obtained using S-4160 microscope (Hitachi, Japan).
Preparation of functionalized MWCNTs
Nitric acid solution (3 mol L−1) was used to functionalize MWCNTs. Briefly, 1.2 g of MWCNTs was mixed with 10 mL of 3 mol L−1 nitric acid solution, and it was refluxed for 20 h at a temperature of 80 °C before the final mixture was filtered, washed several times with water until neutral (pH 7.0), and dried. Significant destruction was observed under acidic treatment when ‒COOH groups were added to the sidewall defects and ends of MWCNTs [18].
Preparation of the DNA-based biosensor
A glassy carbon electrode (GCE) was polished with emery paper and alumina with a particle diameter less than 0.05 mm for 3 min, sonicated in ethanol/water (50:50), eluted with deionized water, and allowed to dry at room temperature. Then, 2.0 mg of MWCNTs was dispersed into 1.0 mL of DMF solvent (2.0 mg mL−1), and the suspension was sonicated for 30 min to get a uniform mixture. Subsequently, 5 μL of the MWCNTs suspension was dropped at the GCE surface and dried at room temperature to obtain MWCNTs/GCE. The MWCNTs/GCE thus obtained was put into PBS (pH 8.0) containing 1.5 mmol L−1l-lysine in order to form PLL/MWCNTs/GCE, which was subjected to scanning by cyclic voltammetry at a scan rate of 50 mV s−1 and in a potential range of − 2.00 to + 2.00 V. To form ds-DNA/PLL/MWCNTs/GCE, ds-DNA was immobilized on the surface of the modified electrode by immersing the PLL/MWCNTs/GCE into 0.1 mol L−1 acetate buffer solution (pH 4.8, 0.002 mol L−1 NaCl) containing 105 mg L−1 ds-DNA at an applied potential of + 0.50 V under stirring (350 rpm) for 600 s. Finally, the unbounded ds-DNA on the biosensor surface was removed by washing the electrode with deionized water for 5 s.
Electrochemical impedance spectroscopy
EIS measurements were taken in the presence of 3.0 mmol L−1 K3[Fe(CN)6]/K4[Fe(CN)6] in 0.1 mol L−1 KCl, as a redox probe, at a polarization potential of 0.18 V and in the frequency range of 0.005 to 105 Hz at an amplitude of 10 mV.
Interaction of diazinon with the ds-DNA
The interaction of diazinon with ds-DNA was investigated by immersing ds-DNA/PLL/MWCNTs/GCE into Tris–HCl buffer solution (pH 7.0) containing different concentrations of diazinon under stirring (350 rpm) for 10 min. Then, the sensor was removed from the solution, and it was washed with the Tris–HCl buffer solution (pH 7.0) to remove any unbonded ds-DNA from ds-DNA/PLL/MWCNTs/GCE. Electrochemical impedance spectra of the biosensor before and after interactions with the diazinon were recorded.
Preparation of real samples
River water was collected from different sites along the Zayandehroud River in Isfahan Province (Iran). Agricultural wastewater was collected from farms located around Isfahan (Iran). Lettuce and tomato were independently squeezed to obtain juices. Then, the whole samples were filtered on Whatman paper No. 45 prior to analysis, which was carried out within the first 48 h after their collection.
Results and discussion
Electropolymerization of l-lysine
PLL/MWCNTs/GCE was prepared by mounting l-lysine layer on MWCNTs/GCE via electropolymerization of l-lysine in PBS (pH 8.0) containing 1.5 × 10−3 mol L−1l-lysine with cyclic voltammetric sweeps in the potential range of − 2.00 to + 2.00 V at a scan rate of 50 mV s−1. As shown in Fig. 1A, during the polymerization, an oxidation current was observed at + 1.60 V, while a reduction peak appeared at −0.70 V in the reverse cathodic scan. The electropolymerization process was performed after eight cycles when the oxidation and reduction peaks’ current did not change. Then, the electrode was rinsed with water to remove any physically adsorbed material before it was dried at ambient temperature. In this way, PLL layers were bonded onto the surface of MWCNTs/GCE [19]. In this work, the choice of the potential ranging from − 2.00 to + 2.00 V was due to the fact that no polymer film was formed at a potential less than + 1.60 V. This could be ascribed to the lack of the free radical at the oxidative potential lower than + 1.60 V. However, extension of the positive potential beyond + 2.00 V would lead to over-oxidation of l-lysine and hence the deterioration of the polymer film. Furthermore, if the reverse scanning final potential was negative than − 2.00 V, gas bubbles were formed, and the polymer film would also be deteriorated [20]. During the electropolymerization of l-lysine at the MWCNTs/GCE, the oxidization of l-lysine monomers leads to the production of α-amino free radicals at a higher positive potential that can link on the electrode surface to form PLL films (Fig. 1B) [20].

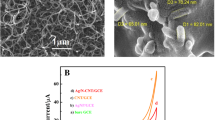
A Electropolymerization of l-lysine in the phosphate buffer solution containing 1.5 mmol L−1 of l-lysine, using cyclic voltammetry at a scan rate of 50 mV s−1; B mechanism of l-lysine electropolymerization on the surface of MWCNTs/GCE; C SEM images of (a) unmodified GCE, (b) PLL/MWCNTs/GCE, and (c) ds-DNA/PLL/MWCNTs/GCE
Characterization of the DNA/PLL/MWCNTs/GCE
The –COOH and –OH groups were formed on the surface of functionalized MWCNTs during the acid treatment. Covalent and noncovalent interactions allow ds-DNA to be immobilized onto the surface of MWCNTs. Nevertheless, the electrostatic repulsion between the negatively charged MWCNTs and DNA surface led to unsatisfactory results [21]. In this study, PLL was used to overcome the problem through electrostatic interaction, whereby the positively charged PLL covered the negatively charged MWCNTs.
SEM is the best tool for investigating the surface morphology of thin layers on the nano- or micro-scales. SEM images of the modified electrodes are shown in Fig. 1C. The surface of the unmodified GCE is shown in Fig. 1C(a). It is clear from the electrode surface morphology that the surface area of the modified electrode is significantly increased as a result if using highly conductive MWCNTs. The homogeneous surface of the modified PLL/MWCNTs/GCE electrode is presented in Fig. 1C(b). Clearly, the positively charged l-lysine was able to cover the whole negatively charged MWCNTs. Therefore, ds-DNA was easily loaded on the uniform surface of PLL/MWCNTs/GCE. The negative charges of the ds-DNA allowed for its proper incorporation into the positively charged surfaces to form the ds-DNA-modified GCE (Fig. 1C(c)).
Cyclic voltammograms (CVs) of the redox probe [Fe(CN)6]3−/4− reveal the characterization of the electrode surface before and after modification. Figure 2a demonstrates the CVs of the bare GCE, PLL/MWCNTs/GCE, and DNA/PLL/MWCNTs/GCE in 5.0 mmol L−1 [Fe(CN)6]3−/4− redox probe containing 0.1 mol L−1 KCl. As can be seen, the anodic and cathodic peak potentials of the redox probe at the surface of the unmodified GCE appeared at + 0.27 and + 0.17 V (vs. Ag/AgCl), respectively [22]. Furthermore, a significant change is observed for the peak currents of Fe(CN) 3−/4−6 at MWCNTs/PLL/GCE as compared to those obtained for the unmodified GCE. This is due to the improved electroactivity of the electrode and the positive charge of the PLL. After immobilization of ds-DNA at the electrode surface, the repulsion between the negative charge of the phosphate group of DNA and Fe(CN) 3−/4−6 led to a decrease in the peak currents of Fe(CN) 3−/4−6 (Fig. 2a).

Cyclic voltammograms (A), and impedance spectra (B) of unmodified GCE (a), MWCNTs/GCE (b), PLL/MWCNTs/GCE (c) and ds-DNA/PLL/MWCNTs/GCE (d) in 5.0 mmol L−1 Fe(CN) 3−/4−6 containing 0.1 mol L−1 KCl. CV measurement at a potential range of − 0.20 to + 0.60 V with a scan rate of 50 mV s−1. Conditions: polarization potential: 0.18 V, frequency: 0.005 to 105 Hz, and potential amplitude of 10 mV
Electrochemical impedance spectroscopy (EIS) was used as a useful technique for investigating the immobilization of ds-DNA on the electrode surfaces. Figure 2b shows a Nyquist plot of the impedance spectrum for GCE under different conditions. In this plot, the electron-transfer-limited procedure at higher frequencies is represented by the semicircle, and the linear part at lower frequencies corresponds to the diffusion process. The increment in the diameter of the semicircle is proportional to the increase in interfacial charge-transfer resistance (Rct). Nanomaterials as modifiers on the GCE surface cause a decline in the surface impedance; in other words, immobilization of the ds-DNA increases the Rct. As shown in Fig. 2b, the Rct of the bare GCE is 300 Ω, while it decreases to 100 Ω after modification of the GCE with MWCNTs and PLL. By ds-DNA immobilization at the surface of the modified electrode, the value of Rct increases to 1200 Ω. This increment in Rct is due to the electrostatic repulsion between the negatively charged ds-DNA surface and the redox probe [Fe(CN)6]3−/4− that inhibits interfacial charge transfer. This phenomenon demonstrates that the ds-DNA was successfully immobilized on the PLL/MWCNTs/GCE surface.
To evaluate the electrochemical surface areas of GCE, MWCNTs/GCE and PLL/MWCNTs/GCE, Fe(CN) 3−6 (1.0 mmol L−1 in 0.10 mmol L−1 KCl) redox probe was used. The microscopic areas of GCE, MWCNTs/GCE and PLL/MWCNTs/GCE were calculated based on the slope of the Ip νs. ν1/2 plot for a known concentration of K3Fe(CN)6 using the Randles–Sevcik equation at a temperature of 25 °C:
where Ip refers to the peak current, A refers to the effective surface area of the electrode, n refers to the electron transfer number, DR refers to the diffusion coefficient, Cs refers to the concentration of K3Fe(CN)6 (1.0 mmol L−1 in 0.1 mol L−1 KCl), and ν is the scan rate. In this case, with n = 1 and DR = 7.6 × 10−6 cm2 s−1. The resultant electrochemical surface areas were obtained as 0.0301, 0.1523, and 0.1323 cm2 for unmodified GCE, MWCNTs/GCE, and PLL/MWCNTs/GCE, respectively.
Optimization of variables
In order to obtain the highest sensitivity for the measurement of diazinon, different parameters such as amounts of MWCNT, PLL, and ds-DNA; the number of cycles required for the polymerization of l-lysine; and immersion time of PLL/MWCNTs/GCE in the ds-DNA solution were optimized using the surface response method. To find their optimum values, a new model was designed and validated using the Minitab software (version 16.2.4). In this model, 33 experiments were designed according to the central composite design (CCD) including 16 cube points, 6 centre points in a cube, 10 axial points, and 1 centre point in the axial. The maximum and minimum levels for each parameter were selected based on a preliminary study using the one-at-a-time method. The model designed in this study was presented as in the following second-order polynomial regression equation:
where Y represents the response; b0 is a constant; b1, b2, b3, b4, and b5 are linear coefficients; b11, b22, b33, b44, and b55 are cross-product coefficients; and b1b2, b1b3, b2b3, b2b4, b3b4, and b4b5 are quadratic coefficients. The coefficients and the standard error of coefficients, as well as the t and p values for the null hypothesis (H0), are reported in Table 1. At the confidence limit of 95%, the parameters with a p value greater than 0.05 do not affect the model and should be removed. The estimated R2 from the model was calculated to be 0.9907, which means that 99% of the data were fitted in the model. Figure 3a, b shows the residual study of the model. As shown in Fig. 3a, the normal probability plot confirms that the variance is normally distributed as well. So, we might proceed assuming that the error terms are normally distributed. Furthermore, the plot of residual versus fitted value indicates that the variance of original observations is constant for all the response values. It was, therefore, concluded that we had a homoscedastic error in the experiment.

a Normal probability plot of the model; b plot of the residual versus fitted value; c surface graphs of the Rct versus different variables
In the final stage, the response surface method was used to calculate the optimum values of the mentioned parameters. The optimum values for MWCNT, ds-DNA, and PLL were obtained at 2 mg mL−1, 100 mg L−1, and 1.5 × 10−3 mol L−1, respectively. Furthermore, the number of cycles and the electrode immersion time in the ds-DNA solution under the optimum conditions were obtained to be eight cycles and 600 s, respectively (Fig. 3c).
Interaction of diazinon with ds-DNA
EIS was used to study the interaction between diazinon and ds-DNA/PLL/MWCNTs/GCE (Fig. 4A). For this purpose, the ds-DNA-modified GCE interacted with different concentrations of diazinon, which led to changes in the interfacial charge-transfer resistance (Rct). The Rct, before and after the interaction with diazinon, was measured. It was observed that Rct decreased with increasing diazinon concentration. Damaging or shielding of the oxidizable groups of guanine and adenine bases led to reducing the Rct during the interaction of diazinon with the ds-DNA.

A: Impedance spectra of ds-DNA/PLL/MWCNTs/GCE in 5.0 mmol L−1 Fe(CN) 3−/4−6 containing 0.1 mol L−1 KCl after its interaction with 0.00 (a), 0.001 (b), 0.050 (c), 0.100 (d), and 5.00 (e) μmol L−1 diazinon; B UV–Vis spectra of 100.0 μmol L−1 ds-DNA before (a) and after (b) interaction with 100.0 μmol L−1 diazinon; and (c) 100.0 μmol L−1 diazinon alone; and C influence of the incubation time on the interaction of diazinon with the DNA at the surface of the biosensor
Absorption spectrophotometry was used to verify the interaction between diazinon and ds-DNA. The absorption spectra of diazinon and ds-DNA before and after interaction with each other are shown in Fig. 4B. Maximum absorbance of diazinon occurred at 225 and 250 nm (Fig. 4B-c) and that of ds-DNA occurred at 295 nm (Fig. 4B-a). The absorption spectra after ds-DNA and diazinon interaction are shown in Fig. 4B-b. The absorbance of ds-DNA at 295 nm decreased after their interaction. The results obtained from absorption spectrophotometry and the reduced impedance spectra are good evidence confirming the interaction between diazinon and ds-DNA [6]. The results of the interaction are a distortion of the DNA because Rct is decreased by increasing the diazinon concentration. After investigation and comparison, the absorption spectra of ds-DNA before and after interaction with diazinon, it was found that the UV–Vis peak position of the ds-DNA moved towards shorter wavelengths (hypsochromic effect) after interaction with diazinon. These variations conclude that in addition to intercalating reactions between diazinon and ds-DNA and opening of ds-DNA helix, the diazinon can induced a significant change in the helical conformation of ds-DNA structure and interacted with ds-DNA in a major groove and an outside binding.
The influence of the incubation time on the interaction of diazinon with the DNA at the surface of the biosensor was studied for 5–25 min. The results of our studied (Fig. 4C) showed that the interaction between the diazinon with the DNA was completed after 10 min. Therefore, 10 min was selected as a suitable interaction time.
Figures of merit
Increasing the concentration of diazinon leads to the destruction of the ds-DNA on the surface of the electrode; hence, the interfacial charge-transfer resistance (Rct) was reduced. Using EIS and tracing the changes in the Rct values after interaction, the biosensor with diazinon at concentrations of (0.001‒0.1) and (0.1‒100) yielded regression equations of R3 = 2.868CDiazinon + 0.1128 (R2 = 0.9981) and R3 = 0.290CDiazinon + 0.6364 (R2 = 0.9964) (Fig. 5), respectively. In these equations, R3 =\(\frac{{R_{2} - R_{1} }}{{R_{1} }}\) (with R1 and R2 representing the values of interfacial charge-transfer resistance (Rct) before and after interaction with diazinon), and C is a diazinon concentration in μmol L−1.

Calibration curve for the determination of diazinon in the concentration range of a 0.001–0.10 and b 0.10–100 μmol L−1 diazinon using ds-DNA/PLL/MWCNTs/GCE under the optimum conditions
In this work, the limit of detection (3 s/m) was estimated to be 0.3 nmol L−1, with s denoting the standard deviation for the determination of blank samples and m designating the slope of the calibration curve. The reproducibility of diazinon measurement as calculated in five consecutive days and repeatability of five diazinon determinations in 1 day were calculated to be 4.3% and 2.8%, respectively (in Tris–HCl buffer solutions (pH 7.0) containing 0.010 μmol L−1 diazinon). Table 2 presents a comparison of the efficiencies obtained in this study and those of previous electrochemical studies in the determination of diazinon.
Interference study
To assay the selectivity of the ds-DNA/PLL/MWCNTs/GCE biosensor for diazinon determination, the effects of the potentially interfering substances commonly found with diazinon in real samples, on the biosensor signal, were investigated. Tolerance limit was defined as the maximum concentration of the interfering substance that caused an error less than 3% in the determination of diazinon concentration under the optimum conditions in the presence of 0.010 μmol L−1 of diazinon. According to the results obtained, 1000-fold of Mn2+, Ca2+, Cu2+, Ni2+, Pb2+, Al3+, Na+, K+, Cl−, CO32−, NO3−, H2PO4−, CH3COO−, IO3−, and SO42− as well as 100-fold of monilate, monolinuron, chlorpyrifos, dichlorvos, parathion, methamidophos, 50-fold of fenitrothion, carbaryl, and amitrole did not affect the biosensor selectivity. These results indicate that the proposed biosensor is selective for diazinon determination.
Real sample analysis
The high sensitivity of the biosensor manufactured in the present study allows for the determination of diazinon in real samples. Environmental water (from the Zayandehroud river), agricultural wastewater, lettuce juice, and tomato juice were used as real samples. Standard addition method was used for measuring diazinon concentrations in the samples. The results for 4-time parallel measurements are given in Table 3. These results confirm the potential of the biosensor for diazinon determination in real samples.
Conclusion
In this study, a novel and highly sensitive ds-DNA biosensor based on multiwall carbon nanotubes and PLL with high selectivity was developed for diazinon determination in real samples. The interaction of diazinon with ds-DNA was transduced via electrochemical and UV–Vis spectroscopy in the first step. The results revealed an interaction between diazinon and ds-DNA. l-lysine as a polycation and small-sized MWCNTs provides a surface with positive charges and a high surface area for the immobilization of ds-DNA. Using the DNA/PLL/MWCNTs/GCE, we were able to monitor the interaction of diazinon with ds-DNA and to determine the trace amounts of diazinon. The observed decrease in the Rct was attributed to the intercalation of diazinon with the ds-DNA on the electrode surface. The proposed biosensor may be recommended as a simple, fast, low-cost, sensitive, and selective tool with low detection limits and a wide linear dynamic range for the detection and determination of diazinon. However, as the interaction between the analyte and DNA is damaging, the sensor cannot regenerate.
References
M. Peyrovi, M.R. Hadjmohammadi, J. Iran. Chem. Soc. 14, 995 (2017)
M. Khadem, F. Faridbod, P. Norouzi, A. Rahimi Foroushani, M.R. Ganjali, S.J. Shahtaheri, J. Iran. Chem. Soc. 13, 2077 (2016)
G. Erdoǧdu, J. Anal. Chem. 58, 562 (2003)
M.A. Mohamed, E.-S.M.E. Mahdy, M.G. Abd-El-Hady, N.M. Ibrahim, H.A. El-Mezayen, M.M.E. Ghanem, Pestic. Biochem. Physiol. 135, 27 (2017)
N. Van Cong, N.T. Phuong, M. Bayley, Ecotoxicol. Environ. Saf. 71, 314 (2008)
M.M. Zahedi, H. Monsef, J. Iran. Chem. Soc. 14, 1099 (2017)
M. Garcés-Garcia, E.M. Brun, R. Puchades, Á. Maquieira, Anal. Chim. Acta 556, 347 (2006)
M.S. Lin, B.I. Jan, H.-J. Leu, J.S. Lin, Anal. Chim. Acta 388, 111 (1999)
Z. Meng, Y. Ma, Microchem. J. 53, 371 (1996)
H. Tavakolia, H. Ghourchian, J. Iran. Chem. Soc. 7, 322 (2010)
M.A. Farajzadeh, A.S. Hojaghan, M.R.A. Mogaddam, J. Food Compos. Anal. 66, 90 (2018)
S. Tursynbolat, Y. Bakytkarim, J. Huang, L. Wang, J. Pharm. Anal. 8, 124 (2018)
E. Mirmomtaz, A.A. Ensafi, S. Soleimanian-Zad, Electrochim. Acta 54, 1141 (2009)
A.A. Ensafi, N. Khademi Farfani, M. Amini, B. Rezaei, J. Iran. Chem. Soc. 14, 1325 (2017)
S. Iijima, Nature 354, 56 (1991)
J. Zhao, C.R. Bradbury, S. Huclova, I. Potapova, M. Carrara, D.J. Ferm’in, J. Phys. Chem. B. 109, 22985 (2005)
J.J. Kakkassery, J.-P. Abid, M. Carrara, D.J. Ferm’in, Faraday Discuss. 125, 157 (2004)
Y. Meng, F. Su, Y. Chen, Tribol. Int. 118, 180 (2018)
Y. Li, H. Wang, B. Yan, H. Zhang, J. Electroanal. Chem. 805, 39 (2017)
C. Jiang, T. Yang, K. Jiao, H. Gao, Electrochim. Acta 53, 2917 (2008)
A. Saljooqi, T. Shamsipur, A. Mostafavi, Bioelectrochemistry 118, 161 (2017)
A.A. Ensafi, M. Amini, B. Rezaei, Colloids Surf. B Biointerfaces 109, 45 (2013)
Y.D. Tanimoto de Albuquerque, L.F. Ferreira, Anal. Chim. Acta 596, 210 (2007)
M. Arvand, M. Dehsaraei, Ionics 24, 2445 (2018)
R.C. Martinez, F.B. Dominguez, J.H. Mendez, Electroanalysis 2, 567 (1990)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zare, A.R., Ensafi, A.A. & Rezaei, B. An impedimetric biosensor based on poly(l-lysine)-decorated multiwall carbon nanotubes for the determination of diazinon in water and fruits. J IRAN CHEM SOC 16, 2777–2785 (2019). https://doi.org/10.1007/s13738-019-01741-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-019-01741-z