
Abstract
A selective, simple and rapid magnetic solid-phase extraction was developed using copper ferrite (CuFe2O4) as an efficient sorbent for the separation and preconcentration of tellurium (IV) ion prior to its determination by electrothermal atomic absorption spectrometry. In this method, only 5 mg of the sorbent was needed to obtain a satisfactory extraction recovery. The CuFe2O4 was synthesized by means of a simple coprecipitation method and subsequently characterized by X-ray diffraction, Fourier transform infrared spectroscopy and scanning electron microscopy. The factors affecting the separation and preconcentration of tellurium (IV) ions including, the type of desorption solvent, desorption solvent concentration and volume, desorption temperature and time, pH, amount of sorbent, extraction temperature and time were investigated and optimized. The effects of interfering ions on the extraction of Te (IV) ions were also investigated. Under the optimized conditions, the method exhibited a linear dynamic range of 0.04–0.5 µg L−1 with good linearity (r2 = 0.9982). The limit of detection and sorption capacity were found to be 0.012 µg L−1 and 89.0 mg g−1, respectively. Enrichment factor was 223. The intraday, interday, and batch-to-batch relative standard deviations were found to be 3.1, 4.4, and 6.0% for 0.25 µg L−1 concentration. Finally, the proposed analytical procedure was successfully applied to monitor tellurium (IV) ions in some aqueous samples, with relative recoveries of > 96%.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tellurium (Te) is a member of the chalcogen elements which is regarded as a rare, non-essential, toxic element [1]. It is used in metallurgy as an additive to steel and copper to provide machinability [2, 3]. It is also used as a coloring agent in chinaware, porcelains, enamels, glasses and ceramic industry. Tellurium has found applications in pharmaceutical and plastic industries and as a catalyst in chemical processes [2, 4].
Tellurium is known to have acute, chronic and teratogenic toxicity [1, 5]. Its acute exposure causes a variety of gastrointestinal and neurological symptoms [6], can accumulate in the kidney, heart, liver and spleen [2], and induce the degeneracy of liver and kidney in excess of 0.002 g kg−1 [7]. After microbial action, it can be transformed into volatile, organometalloid compounds [8] with the consequent modification of its transport pattern and toxicological behavior [1]. However, its bioavailability and environmental transport mechanism highly depend on its oxidation state. Te (IV) ion is 10 times more toxic than Te (VI) ion [3].
Determination methods of tellurium ions include, inductively coupled plasma optical emission spectrometry (ICP-OES) [9], hydride generation microwave-induced plasma atomic emission spectrometry (HG-MIP-AES) [10, 11], inductively coupled plasma mass spectrometry (ICP-MS) [12,13,14], spectrophotometry [15, 16], hydride generation atomic fluorescence spectrometry (HG-AFS) [17], hydride generation atomic absorption spectrometry (HG-AAS) [18,19,20], electrothermal atomic absorption spectrometry (ET-AAS) [21,22,23], and electrochemical techniques [24, 25]. Considering the high cost of ICP-OES and ICP-MS, atomic absorption spectroscopy is still widely used. Graphite furnace atomic absorption spectrometry (GFAAS) is a very rapid and convenient method for determination of Te ions in various samples.
Since the tellurium ions’ concentrations in environmental and industrial samples are usually far below the µg L−1 levels [26], preconcentration and separation procedures are usually necessary prior to the analysis. The most commonly used separation and enrichment techniques is liquid–liquid extraction (LLE) [27, 28], which consumes large volumes of hazardous solvents and is laborious and time-consuming [29]. Dispersive liquid–liquid microextraction (DLLME) [21] and hollow fiber liquid phase microextraction (HF-LPME) [22] have been introduced as alternatives to conventional LLE. Solid-phase extraction (SPE) has also been used for the separation and preconcentration of Te ions [9, 12, 13, 18, 26] with a variety of sorbents.
Magnetic nanoparticles (MNPs), as a new type of SPE sorbent, have attracted increasing attention [30,31,32]. Magnetic solid-phase extraction (MSPE) has now become one of the most common strategies for separation and preconcentration of analytes. This technique of separation was first reported by Robinson et al. [33], but the term magnetic solid-phase extraction was first introduced by Sajarikova and Safarik [34]. This technique is very convenient, since the magnetic sorbent can be recovered from the sample solution under an external magnetic field. The main advantages of MSPE include simplicity, low cost, rapidity, lower solvent consumption, high enrichment factor and the ability to couple with different detection methods [35]. In this method, separation and collection are easier and faster than conventional SPE, avoiding common problems such as channeling or blocking of cartridges or disks.
A suitable sorbent for the MSPE technique should be stable and have quantitative sorption capacity [36]. Ferrite nanoparticles with the general formula MFe2O4 (M = Cu, Mn, Mg, Zn, Ni, Co, and other metals) can be easily synthesized and functionalized using a wide range of techniques [37]. Their favorable properties include high absorption capacity [38, 39], hardness, high coercivity, moderate magnetization, and high physical, chemical and corrosive stability [40]. Among ferrites, copper ferrite (CuFe2O4) is one of the most important ferrites with interesting properties and potential applications. CuFe2O4 is known to exist in tetragonal and cubic structures and generally has an inverse spinel structure [41]. Using copper in ferrite structure has the advantage of being less toxic compared to other toxic metals.
In this paper, CuFe2O4 nanoparticles were synthesized through coprecipitation method and characterized by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR) and scanning electron microscopy (SEM). Eventually, its capability as a sorbent for the extraction of tellurium (IV) ions from aqueous samples was examined, and a ligandless procedure based on magnetic solid-phase extraction prior to its determination by ET-AAS was developed.
Experimental
Materials and reagents
All reagents used were of analytical-reagent grade and de-ionized water was used throughout the work. Tellurium (IV) ion working solutions were prepared by appropriate dilutions of a 1000 mg L−1 AAS standard solution purchased from Fluka (Switzerland).
Instrumentation
A Varian Spectra model AA 220 atomic absorption spectrometer (Melbourne, Australia) with a graphite furnace atomizer equipped with deuterium background correction was used for determination of tellurium (IV) ions. The temperature program for the graphite furnace was optimized (Online Resource 1). The optimum temperature program was: ashing 1000 °C (10 s), atomization 2100 °C (2 s). A tellurium hollow cathode lamp was used as the light source operated at 10 mA and 214.3 nm. Samples were introduced in a co-injection mode of 30 µL tellurium (IV) ion solution and 10 µL 5% Ni (NO3)2 as chemical modifier. The FT-IR instrument was a Tensor 27 FT-IR spectrometer from Brucker (Ettlingen, Germany). The morphology of the sorbent was determined by KYKY-EM3200 scanning electron microscope (Beijing, China) operated at 26 kV. X-ray diffraction pattern of copper ferrite nanoparticles was obtained on a Panalytical powder X-ray diffraction system model X’PertPRO (Almelo, Netherlands) at a tube voltage of 40 kV and a tube current of 30 mA using a copper cathode as the X-ray source (λ = 1.542 Å). A Metrohm digital pH-meter model 713 (Herisau, Switzerland) was used for pH measurements. A Bandelin Sonorex Super RK 225 ultrasonic bath at 35 kHz (Berlin, Germany) was used for the extraction procedure. Magnetic separations were carried out by a Nd–Fe–B supermagnet with 1.2 T magnetic field. A WF binder oven model 7200 (Tuttlingen, Germany) and a Lenton furnace model EF 11/8B (Hope Valley, England) were used for synthesis purposes. All containers and glassware used in the extraction studies were soaked in 3 mol L−1 nitric acid overnight and rinsed with deionized water before use.
Synthesis of copper ferrite magnetic nanoparticles
The MNPs were produced by coprecipitation method according to the procedure reported by Stefanova et al. with some modifications [42]. The synthesis was performed by rapidly adding 350 mL boiling solution of 0.25 mol L−1 NaOH to the precursor solution containing 0.017 mol L−1 Cu2+ and 0.034 mol L−1 Fe3+ in a volume of 500 mL heated to 30 °C. To avoid simultaneous formation of non-magnetic and paramagnetic particles at higher pH values, the final pH of the coprecipitating mixture was adjusted at ~ 8, if necessary. The precipitation process was continued for 3 h after increasing the temperature to 80 °C.
After completion of precipitation of nanoparticles, the MNPs were separated from the supernatant solution by means of the supermagnet and washed with deionized water until the pH of the washing solution reached a value of 7.0, then washed with ~ 50 mL of ethanol and the final product was dried for 24 h at 65 °C.
To prepare monodisperse CuFe2O4 nanoparticles, the powder was ultrasonically dispersed in ethanol, then centrifuged, rewashed with ethanol and dried [43]. Then, the dried powder was annealed in air for 3 h at 600 °C.
MSPE procedure
Extraction procedure consisted of the following steps: a 100 mL portion of sample solution containing Te (IV) ions (0.4 µg L−1) was placed into a 100 mL beaker. An optimum amount of CuFe2O4 (5 mg) was added and the mixture was stirred for 20 min on a heater-stirrer at 40 °C. Then, the sorbent was magnetically separated using a supermagnet placed outside the beaker. Two portions of 10 mL distilled water were used to wash the sorbent from non-adsorbed Te ions, and then the supernatant solution was decanted. Now, 0.5 mL of 7 mol L−1 HNO3 was added and stirred for 10 min at 55 °C to elute the extracted Te ions from the nanoparticles. After separating the nanoparticles, the remaining solution was evaporated under a gentle stream of nitrogen. Now, the dried residue was redissolved in 100 µL of 1 mol L−1 HNO3 by ultrasonication for 1 min. The resultant solution was analyzed by ET-AAS. All above operations were carried out in triplicate.
Real sample
Water samples, including tap water, well water and seawater were filtered through a 0.45 µm Millipore filter (Schwalbach, Germany) and subjected to the MSPE-ET-AAS.
Results and discussion
Characterization of copper ferrite
The surface morphology, structure and the size of synthesized nanoparticles were characterized by SEM. The SEM image shown in Fig. 1a revealed that the size of the synthesized nanoparticles produced was between 40 and 60 nm.

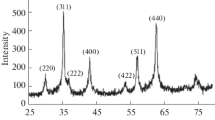
Scanning electron microscope image (a), FT-IR spectrum (b) and X-ray diffraction pattern (c) of copper ferrite nanoparticles
The FT-IR spectrum of the copper ferrite nanoparticles is shown in Fig. 1b. The structure of copper ferrite nanoparticles was confirmed by the presence of Fe–O and Cu–O stretching vibrations at 575 cm−1 and 417 cm−1, respectively, the broad stretching band at 3426 cm−1 for H2O molecules and surface OH groups, and the band around 1600 cm−1 corresponding to the bending mode of H2O molecules [44, 45].
The identity of the copper ferrite nanoparticles was verified by comparing its XRD pattern with that of cuprospinel copper iron oxide having a cubic crystal system (Fig. 1c). The mean particle diameter found was 12.1 nm calculated from the XRD pattern using the Debye–Scherrer equation. The particle dimension obtained by SEM, however, is higher than the crystalline size obtained by XRD pattern (Fig. 1a). This difference may be explained due to the presence of aggregates consisting of several crystallites and/or poor crystallinity. Seven characteristic diffraction peaks at 2θ values of 18.57°, 30.52°, 35.85°, 39.17°, 43.71°, 57.50°, 62.61° can be indexed, respectively, to the (111), (220), (311), (222), (400), (511), and (440) planes of the copper iron oxide nanoparticles with a cubic structure.
Optimization of the extraction parameters
To take full advantage of the procedure, the extraction conditions must be optimized. Various experimental parameters were studied to optimize the system. A one-at-a-time strategy was used for this purpose. The initial values of extraction parameters were: 1 mL HNO3 3 mol L−1, pH 6.0, 3 mg sorbent, at 25 °C, 10 min extraction time and 10 min desorption time.
Sample pH
The pH of the sample solution is another significant variable that controls the extraction of Te (IV) ions by the sorbent. An appropriate pH is not only important for the extraction of the element but also important for the selectivity behavior. The solution pH can affect both aqueous chemistry and surface-binding sites of the sorbent. Tellurium is rather an amphoteric element: it can enter into solution in the form of both cations and anions [15, 46, 47].
Point of zero charge (pHpzc) is a useful factor in sorption studies. This allows one to hypothesize on the ionization of functional groups and their interactions with analytes. The point of zero charge for CuFe2O4 is 7.3 [48, 49]. This means that the surface of the nanoparticles is protonated and positively charged at pHs below 7.3. As the pH of the solution is increased above this value, the surface becomes negatively charged due to deprotonation reaction.
In this work, the effect of pH was investigated in the pH range of 2–12 (Fig. 2a). It can be seen that the extraction of Te (IV) ions increases with increasing pH of the solution to reach a maximum at pH 6.0. The sorption curve of Te (IV) ions onto CuFe2O4 nanoparticles can be divided into two regions. In the first region (pH 2.0–6.0), the sorption efficiency of Te (IV) ions gradually increases when the pH increases from 2.0 to 6.0. When the pH is low, the surface of CuFe2O4 nanoparticles is positively charged. Under this condition, an electrostatic repulsion force will exist between the positive tellurium ions and CuFe2O4 nanoparticles, preventing sorption [50]. As pH increases, the surface of CuFe2O4 nanoparticles became less positive. At the same time, the concentration of tellurium anions (HTeO3−) increases, leading to a decrease in electrostatic repulsion force which will facilitate the sorption of Te (IV) ions onto CuFe2O4 nanoparticles. In the second region (pH 6–12), the removal of Te (IV) ions decreases along with the increase in pH. In this region, the sorbent surface gradually becomes negatively charged and electrostatic repulsion between the dominating negatively charged Te species and the sorbent surface decreases Te (IV) uptake.

Effect of pH (a), sorbent amount (b), extraction temperature (c) and extraction time (d). Extraction conditions: a ext. time 10 min; ext. temp. 25 °C; desorp. time 10 min; desorp. temp. 25 °C; desorp. solvent 1.0 mL 3 mol L−1 HNO3; sorbent amount 3 mg. b Similar to a except pH 6.0. c Similar to a except pH 6.0; sorbent amount 5 mg. d Similar to a except pH 6.0; sorbent amount 5 mg; ext. temp. 40 °C
Amount of sorbent
The amount of sorbent is one of the main factors affecting the extraction of analytes in MSPE. Hence, the amount of sorbent varied between 1 and 9 mg (Fig. 2b). As seen, maximum sorption occurs at 5 mg of sorbent. Therefore, 5 mg of sorbent was used in all subsequent experiments.
Extraction temperature and time
In extraction procedures, it is desirable to have the shortest equilibration time and lowest possible equilibration temperature beside efficient extraction. Therefore, the dependence of extraction efficiency upon extraction temperature and time were studied at 10–70 °C and 5–30 min, respectively. The results showed that 40 °C and 20 min were adequate to achieve a maximum extraction efficiency (Fig. 2c, d). Temperature mainly influences metal ion adsorption by affecting the chemical structure of an adsorbent surface, as well as the physical and chemical status of a solution. The results show that a better adsorption performance could be obtained at a higher temperature and higher temperature facilitates the adsorption of metal ions. Therefore, the adsorption of Te (IV) ions onto copper ferrite nanoparticles is endothermic. This may result in increasing the mobility of metal ions and number of active sites on CuFe2O4 for the adsorption with increased temperature [45].
Desorption solvent
To find a suitable desorption solvent, HNO3, HCl, H2SO4, CH3COOH and thiourea solutions were examined. From these solvents, thiourea, HCl and H2SO4 were not suitable because thiourea separated during solvent evaporation and HCl and H2SO4 created high background signals during atomic absorption measurements. From HNO3 and CH3COOH, the former was found to be a better desorption solvent of tellurium (IV) ions from the sorbent (Fig. 3a). Subsequently, the concentration of HNO3 varied from 1 mol L−1 to concentrated nitric acid. It was found that the highest signal was obtained at 7 mol L−1 HNO3. At higher than 7 mol L−1 concentrations, background signal increased. Therefore, 7 mol L−1 HNO3 was select as the optimum concentration of desorption solvent (Fig. 3b).

Effect of type (a), concentration (b) and volume (c) of desorption solvent. Extraction conditions: a pH 6.0; ext. time 20 min; ext. temp. 40 °C; desorp. time 10 min; desorp. temp. 25 °C; solvent vol. 1.0 mL. b Similar to a except desorp. solvent HNO3. c Similar to a except desorp. solvent HNO3; desorp. solvent conc. 7 mol L−1
The effect of the volume of desorption solvent on the signal was also studied by varying the volume in the range of 0.1–1 mL. The results indicated that 0.5 mL HNO3 was sufficient for complete desorption of tellurium (IV) ions (Fig. 3c). Thus, 0.5 mL of 7 mol L−1 HNO3 was employed in further experiments.
Desorption temperature and time
To achieve complete desorption, the effect of desorption temperature and time on the extraction efficiency of tellurium (IV) ions were investigated. As shown in Fig. 4a, b, when the desorption temperature and time were increased to 55 °C and 10 min, respectively, absorbance signals reached their maxima. Therefore, 55 °C and 10 min were used for desorption of tellurium (IV) ions in further experiments.

Effect of desorption temperature (a) and desorption time (b). Extraction conditions: a pH 6.0; ext. time 20 min; ext. temp. 40 °C; desorp. time 10 min; desorp. solvent. 0.5 mL 7 mol L−1 HNO3; sorbent amount 5 mg. b similar to a except desorp. temp. 55 °C
Sorbent capacity and enrichment factor
The sorbent capacity, i.e., the maximum amount of analyte retained per gram of sorbent, was determined by dispersing 5 mg of copper ferrite (CuFe2O4) in 100 mL of 0.5, 1.5, 2.5, 3.5, 4.5, 4.6, 4.7 mg L−1 Te (IV) ions under the aforementioned optimized conditions. Then, the amount of Te (IV) ions remaining in the solution was measured and the sorbent capacity was calculated using Eq. (1):
where qe is sorbent capacity (mg g−1), Ci and Ce are the initial and equilibrium concentrations (mg L−1) of tellurium (IV) ions, respectively; W (g) and V (L) are the weight of sorbent and the initial volume of aqueous solution, respectively. The adsorption isotherm was of Langmuir model and the maximum sorbent capacity for Te (IV) ions was 89 mg g−1 of sorbent.
The enrichment factor was calculated as the ratio of the slope of the calibration curve obtained from the preconcentrated samples (m) to that obtained without preconcentration (m0) as shown in Eq. (2):
The enrichment factor for the process was found to be 223.
Interference study
To assess the analytical application of the present procedure, the effects of various foreign ions were examined at optimized conditions described above. For this purpose, 0.25 µg L−1 solutions of Te (IV) ions containing various amounts of an interfering ion were prepared and analyzed. An ion was considered to interfere when its presence produced a relative error of more than 5% in absorbance. The results obtained (Table 1) show how selective the method is, and that the ions studied had no obvious influence on the analyte signal up to at least a 500 molar ratio. These results permit the application of the proposed system to interference-free determination of trace tellurium (IV) ions in real samples.
Analytical performance
Under optimized conditions, the analytical performance of the proposed method was evaluated. The calibration curve constructed using eight different concentrations was linear in the range of 0.04–0.5 µg L−1. The equation for the regression line was A = 2.2569 (± 0.0336) C + 0.0398 (± 0.0080), where A is the absorbance (peak height) and C is initial concentration of tellurium (IV) ions in µg L−1. The coefficient of determination (r2) for the regression line was 0.9982. The limits of detection (LOD) and quantification (LOQ) for ten replicate measurements on the blank sample and, respectively, defined as 3Sb/m and 10Sb/m, were 0.012 and 0.040 µg L−1. The intraday relative standard deviation (RSD%) of the method was determined through five replicate measurements of Te (IV) ions at 0.25 µg L−1 and was found to be 3.1%. The interday relative standard deviation of the method for three consecutive days was 4.4%. The sorbent-to-sorbent RSD% was also studied and found to be 6.0% (n = 5).
To evaluate the efficiency of the synthesized sorbent in extraction and preconcentration of Te (IV) ions, the developed method was used to determine Te (IV) ions in real samples. The results, along with the relative recoveries for the spiked samples, are given in Table 2. As shown, the relative recoveries found are > 96%. The results showed that the spiked amount could be quantitatively recovered from the samples.
A comparison of the proposed method with some other recent reported methods in determination of Te (IV) ions can be found in Table 3. As seen, the linear dynamic range (LDR), linearity and precision of the present method are comparable to the literature data and the LOD is better than most of them.
Conclusions
In this study, bare copper ferrite nanoparticles have been used for MSPE operations for the first time. This method offers high sorption capacity and fast sorption/desorption kinetic for the separation and preconcentration of Te (IV) ions from aqueous solutions before determination by ET-AAS. The synthesis of the proposed magnetic sorbent is simple and inexpensive. The synthesized copper ferrite was characterized by FT-IR, SEM and XRD showing the nanometer size nature of the sorbent. The isolation of the sorbent from sample solutions is extremely easy, since a simple magnet is required to separate it from the sample matrix. The proposed analytical method gives a low limit of detection, good precision and accuracy, almost complete recovery and high enrichment factor. The solvent-free nature, ease of operation, high selectivity and sensitivity are other advantages of this method.
References
L.A. Ba, M. Döring, V. Jamier, C. Jacob, Tellurium: an element with great biological potency and potential. Org. Biomol. Chem. 8(19), 4203–4216 (2010)
L. Gerhardsson, in Handbook on the Toxicology of Metals, ed. by G.F. Nordberg, B.A. Fowler, M. Nordberg, L. Friberg, 3rd edn. (Academic Press, Burlington, 2007), p. 815
H.A. Schroeder, J. Buckman, J.J. Balassa, Abnormal trace elements in man: tellurium. J. Chron. Dis. 20(3), 147–161 (1967)
A. Naumov, Selenium and tellurium: state of the markets, the crisis, and its consequences. Metallurgist 54(3–4), 197–200 (2010)
T. Sadeh, in Chemistry of Organic Selenium and Tellurium Compunds, vol. 2, ed. by S. Patai (Wiley, Chichester, 1987), p. 367
A. Larner, Alzheimer’s disease, Kuf’s disease, tellurium and selenium. Med. Hypotheses 47(2), 73–75 (1996)
Z. Chai, H. Zhu, Introduction to trace element chemistry (Chinese Atomic Energy Press, Beijing, 1994), pp. 201–204
T.G. Chasteen, R. Bentley, Biomethylation of selenium and tellurium: microorganisms and plants. Chem. Rev. 103(1), 1–26 (2003)
M. Kaplan, S. Cerutti, S. Moyano, R. Olsina, L. Martinez, J. Gásquez, On-line preconcentration system by coprecipitation with lanthanum hydroxide using packed-bed filter for the determination of tellurium in water by ICP-OES with USN. Inst. Sci. Technol. 32(4), 423–431 (2004)
A. Matsumoto, T. Shiozaki, T. Nakahara, Simultaneous determination of bismuth and tellurium in steels by high power nitrogen microwave induced plasma atomic emission spectrometry coupled with the hydride generation technique. Anal. Bioanal. Chem. 379(1), 90–95 (2004)
A. Matsumoto, A. Oheda, T. Nakahara, in Determination of Tellurium in Steels by High Power Nitrogen Microwave Induced Plasma Atomic Emission Spectrometry Coupled with Hydride Generation Technique, Analytical Sciences/Supplements Proceedings of IUPAC International Congress on Analytical Sciences 2001 (ICAS 2001), The Japan Society for Analytical Chemistry, (2002) pp. i963–i966
C. Yu, Q. Cai, Z.-X. Guo, Z. Yang, S.B. Khoo, Simultaneous speciation of inorganic selenium and tellurium by inductively coupled plasma mass spectrometry following selective solid-phase extraction separation. J. Anal. Atom. Spectrom. 19(3), 410–413 (2004)
K. Urbánková, M. Moos, J. Machát, L. Sommer, Simultaneous determination of inorganic arsenic, antimony, selenium and tellurium by ICP-MS in environmental waters using SPE preconcentration on modified silica. Int. J. Environ Anal. Chem. 91(11), 1077–1087 (2011)
C. Huang, B. Hu, Speciation of inorganic tellurium from seawater by ICP-MS following magnetic SPE separation and preconcentration. J. Sep. Sci. 31(4), 760–767 (2008)
L. Zhang, M. Zhang, X. Guo, X. Liu, P. Kang, X. Chen, Sorption characteristics and separation of tellurium ions from aqueous solutions using nano-TiO2. Talanta 83(2), 344–350 (2010)
H. Khajehsharifi, M. Mousavi, J. Ghasemi, M. Shamsipur, Kinetic spectrophotometric method for simultaneous determination of selenium and tellurium using partial least squares calibration. Anal. Chim. Acta 512(2), 369–373 (2004)
E. Ródenas-Torralba, P. Cava-Montesinos, Á Morales-Rubio, M.L. Cervera, M. de la Guardia, Multicommutation as an environmentally friendly analytical tool in the hydride generation atomic fluorescence determination of tellurium in milk. Anal. Bioanal. Chem. 379(1), 83–89 (2004)
A. Körez, A.E. Eroğlu, M. Volkan, O.Y. Ataman, Speciation and preconcentration of inorganic tellurium from waters using a mercaptosilica microcolumn and determination by hydride generation atomic absorption spectrometry. J. Anal. Atom. Spectrom. 15(12), 1599–1605 (2000)
M. Xi, R. Liu, P. Wu, K. Xu, X. Hou, Y. Lv, Atomic absorption spectrometric determination of trace tellurium after hydride trapping on platinum-coated tungsten coil. Microchem. J. 95(2), 320–325 (2010)
H. Matusiewicz, M. Krawczyk, Determination of tellurium by hydride generation with in situ trapping flame atomic absorption spectrometry. Spectrochim. Acta Part B 62(3), 309–316 (2007)
N.M. Najafi, H. Tavakoli, R. Alizadeh, S. Seidi, Speciation and determination of ultra trace amounts of inorganic tellurium in environmental water samples by dispersive liquid–liquid microextraction and electrothermal atomic absorption spectrometry. Anal. Chim. Acta 670(1–2), 18–23 (2010)
E. Ghasemi, N.M. Najafi, F. Raofie, A. Ghassempour, Simultaneous speciation and preconcentration of ultra traces of inorganic tellurium and selenium in environmental samples by hollow fiber liquid phase microextraction prior to electrothermal atomic absorption spectroscopy determination. J. Hazard. Mater. 181(1–3), 491–496 (2010)
E. Ghasemi, N.M. Najafi, S. Seidi, F. Raofie, A. Ghassempour, Speciation and determination of trace inorganic tellurium in environmental samples by electrodeposition-electrothermal atomic absorption spectroscopy. J. Anal. Atom. Spectrom. 24(10), 1446–1451 (2009)
P. Sharma, A. Agarwal, Voltammetric ultra trace determination of tellurium. Anal. Lett. 39(7), 1421–1427 (2006)
P. Zong, Y. Nagaosa, Cathodic stripping voltammetric determination of tellurium (IV) with in situ plated bismuth-film electrode. Anal. Lett. 42(13), 1997–2010 (2009)
J. Pedro, J. Stripekis, A. Bonivardi, M. Tudino, Determination of tellurium at ultra-trace levels in drinking water by on-line solid phase extraction coupled to graphite furnace atomic absorption spectrometer. Spectrochim. Acta, Part B 63(1), 86–91 (2008)
I.S. Balogh, V. Andruch, Comparative spectrophotometric study of the complexation and extraction of tellurium with various halide ions and N, N′-di (acetoxyethyl) indocarbocyanine. Anal. Chim. Acta 386(1–2), 161–167 (1999)
C.-H. Chung, E. Iwamoto, M. Yamamoto, Y. Yamamoto, Selective determination of arsenic (III, V), antimony (III, V), selenium (IV, VI) and tellurium (IV, VI) by extraction and graphite furnace atomic absorption spectrometry. Spectrochim. Acta Part B 39(2–3), 459–466 (1984)
S. Garboś, E. Bulska, A. Hulanicki, Z. Fijałek, K. Sołtyk, Determination of total antimony and antimony (V) by inductively coupled plasma mass spectrometry after selective separation of antimony (III) by solvent extraction with N-benzoyl-N-phenylhydroxylamine. Spectrochim Acta Part B 55(7), 795–802 (2000)
M.A. Karimi, A. Hatefi-Mehrjardi, S.Z. Mohammadi, A. Mohadesi, M. Mazloum-Ardakani, M.R.H. Nezhad, A.A. Kabir, Solid phase extraction of trace amounts of Pb (II) in opium, heroin, lipstick, plants and water samples using modified magnetite nanoparticles prior to its atomic absorption determination. J. Iran. Chem. Soc. 9(2), 171–180 (2012)
H. Eskandari, A. Bezaatpour, F. Eslami, Naked magnetite nanoparticles for both clean-up and solid-phase extraction-trace determination of mercury. J. Iran. Chem. Soc. 14(2), 457–469 (2017)
S. Seidi, M. Majd, Polyaniline-functionalized magnetic graphene oxide for dispersive solid-phase extraction of Cr (VI) from environmental waters followed by graphite furnace atomic absorption spectrometry. J. Iran. Chem. Soc. 14(6), 1195–1206 (2017)
P. Robinson, P. Dunnill, M. Lilly, The properties of magnetic supports in relation to immobilized enzyme reactors. Biotechnol. Bioeng. 15(3), 603–606 (1973)
M. Šafaříková, I. Šafařík, Magnetic solid-phase extraction. J. Magn. Magn. Mater. 194(1), 108–112 (1999)
M.H. Mashhadizadeh, M. Amoli-Diva, M.R. Shapouri, H. Afruzi, Solid phase extraction of trace amounts of silver, cadmium, copper, mercury, and lead in various food samples based on ethylene glycol bis-mercaptoacetate modified 3-(trimethoxysilyl)-1-propanethiol coated Fe3O4 nanoparticles. Food Chem 151, 300–305 (2014)
C. Huang, B. Hu, Silica-coated magnetic nanoparticles modified with γ-mercaptopropyltrimethoxysilane for fast and selective solid phase extraction of trace amounts of Cd, Cu, Hg, and Pb in environmental and biological samples prior to their determination by inductively coupled plasma mass spectrometry. Spectrochim. Acta Part B 63(3), 437–444 (2008)
A. Subha, M.G. Shalini, S.C. Sahoo, in Magnetic Studies of CuFe 2 O 4 Nanoparticles Prepared by Co-precipitation Method, AIP Conference Proceedings (AIP Publishing, 2016), p. 020416
J. Zhang, M. Li, Y. Li, Z. Li, F. Wang, Q. Li, W. Zhou, R. Lu, H. Gao, Application of ionic-liquid-supported magnetic dispersive solid-phase microextraction for the determination of acaricides in fruit juice samples. J. Sep. Sci. 36(19), 3249–3255 (2013)
A.A. Ensafi, S. Rabiei, B. Rezaei, A.R. Allafchian, Magnetic solid-phase extraction to preconcentrate ultra trace amounts of lead (ii) using modified-carbon nanotubes decorated with NiFe2O4 magnetic nanoparticles. Anal. Methods 5(16), 3903–3908 (2013)
K. Maaz, A. Mumtaz, S. Hasanain, A. Ceylan, Synthesis and magnetic properties of cobalt ferrite (CoFe2O4) nanoparticles prepared by wet chemical route. J. Magn. Magn. Mater. 308(2), 289–295 (2007)
N.K. Thanh, N.P. Duong, D.Q. Hung, T.T. Loan, T.D. Hien, Structural and magnetic characterization of copper ferrites prepared by using spray co-precipitation method. J. Nanosci. Nanotechnol. 16(8), 7949–7954 (2016)
V. Stefanova, D. Georgieva, V. Kmetov, I. Roman, A. Canals, Unmodified manganese ferrite nanoparticles as a new sorbent for solid-phase extraction of trace metal–APDC complexes followed by inductively coupled plasma mass spectrometry analysis. J. Anal. At. Spectrom. 27(10), 1743–1752 (2012)
Z. Zi, Y. Sun, X. Zhu, Z. Yang, J. Dai, W. Song, Synthesis and magnetic properties of CoFe2O4 ferrite nanoparticles. J. Magn. Magn. Mater. 321(9), 1251–1255 (2009)
M. Gholinejad, B. Karimi, F. Mansouri, Synthesis and characterization of magnetic copper ferrite nanoparticles and their catalytic performance in one-pot odorless carbon-sulfur bond formation reactions. J. Mol. Catal. A Chem. 386, 20–27 (2014)
F. Talebzadeh, R. Zandipak, S. Sobhanardakani, CeO2 nanoparticles supported on CuFe2O4 nanofibers as novel adsorbent for removal of Pb (II), Ni (II), and V (V) ions from petrochemical wastewater. Desal. Water Treat 57(58), 28363–28377 (2016)
M. Bouroushian, Electrochemistry of the Chalcogens, Electrochemistry of Metal Chalcogenides, (Springer, Berlin, 2010), pp. 57–75
E. Rudnik, P. Biskup, Electrochemical behavior of tellurium in acidic nitrate solutions. Metall. Foundry Eng. 40, 15–31 (2014)
Y.-J. Tu, C.-F. You, C.-K. Chang, S.-L. Wang, T.-S. Chan, Arsenate adsorption from water using a novel fabricated copper ferrite. Chem. Eng. J. 198, 440–448 (2012)
Y.-J. Tu, C.-F. You, C.-K. Chang, S.-L. Wang, T.-S. Chan, Adsorption behavior of As (III) onto a copper ferrite generated from printed circuit board industry. Chem. Eng. J. 225, 433–439 (2013)
P.V. Grundler, J. Brugger, B.E. Etschmann, L. Helm, W. Liu, P.G. Spry, Y. Tian, D. Testemale, A. Pring, Speciation of aqueous tellurium (IV) in hydrothermal solutions and vapors, and the role of oxidized tellurium species in Te transport and gold deposition. Geochim. Cosmochim. Acta 120, 298–325 (2013)
J. Pedro, J. Stripekis, A. Bonivardi, M. Tudino, Thermal stabilization of tellurium in mineral acids solutions: use of permanent modifiers for its determination in sulfur by GFAAS. Talanta 69(1), 199–203 (2006)
L. Luo, Y. Tang, M. Xi, W. Li, Y. Lv, K. Xu, Hydride generation induced chemiluminescence for the determination of tellurium (IV). Microchem. J. 98(1), 51–55 (2011)
P. Wu, Y. Zhang, R. Liu, Y. Lv, X. Hou, Comparison of tungsten coil electrothermal vaporization and thermospray sample introduction methods for flame furnace atomic absorption spectrometry. Talanta 77(5), 1778–1782 (2009)
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Narimani-Sabegh, S., Noroozian, E. Magnetic solid-phase extraction with copper ferrite nanoparticles for the separation and preconcentration of ultra-trace amounts of tellurium (IV) ion in aqueous samples. J IRAN CHEM SOC 16, 73–81 (2019). https://doi.org/10.1007/s13738-018-1482-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-018-1482-0