
Abstract
The [Co(5-XSalen)(PEt3)(H2O)]ClO4 (where Salen = bis(salicylaldehyde)1,2-ethylenediamine and X = H, MeO, NO2, Br) complexes were successfully synthesized and characterized by different techniques such as FT-IR, UV–Vis, 1HNMR, 13CNMR and 31PNMR. The coordination geometry of the [Co(5-BrSalen)(PEt3)(H2O)]ClO4 complex was determined by X-ray crystallography. The complex has six-coordinated pseudo-octahedral geometry in which the O(1), O(2), N(1) and N(2) atoms of the Schiff base form the equatorial plane. The cyclovoltammetry was used to study the electrochemical properties of cobalt complexes, and the results reveal the anodic peak potential becomes more positive in order to MeO < H < Br < NO2. DFT calculations were also done to investigate structures, electronic spectra and infrared spectra of the complexes. The synthesized complexes [Co(Salen)(PEt3)(H2O)]ClO4, [Co(Salen)(PBu3)(H2O)]ClO4 and [Co(5-NO2Salen)(PEt3)(H2O)]ClO4 were incorporated within Montmorillonite-K10 (MMT) nanoclay. Furthermore, structural, thermal and morphological properties of the prepared nanohybrids were verified by FT-IR, XRD, TGA-DTG, EDX, SEM and TEM techniques. XRD results of the new nanohybrid materials elucidate that the Schiff base complexes were placed at the most outerlayer spaces of MMT clay.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Biological activities and chelating agents capability of Schiff bases make them as an important and interesting type of ligands, especially in application as models for biological systems [1,2,3]. Schiff base complexes show a good stability in different oxidation states and have been applied in a wide range of application, e.g., catalysts in organic redox and electrochemical reduction [4,5,6,7]. Furthermore, these types of compounds can be applied as catalysts in some important chemical reactions such as alkenes hydrogenation [8, 9], amino group’s transformation [10] and oxidation of alcohols [11, 12].
Cobalt complexes prepared from Schiff base ligands have been studied as models for the cobalamin (B12) coenzymes [13] and used as an oxygen carrier [14]. Furthermore, catalytic activity of these series of compounds was studied on the preliminary oxygenation of phenols [15] and amines [16]. The thermodynamic and redox properties of [CoL(PR3)(H2O)[+ (L = N2O2 Schiff base) compounds were studied previously [17,18,19,20,21,22,23]. Recently, the interaction of cobalt Schiff base complexes and MMT-K10 was studied [24, 25]. Moreover, heterogeneous catalytic activity of incorporated palladium compounds onto MMT-K10 was reported [26].
The Schiff bases with general formula of 5-XSalen [where Salen = bis (salicylaldehyde) 1,2-ethylenediamine and X = H, NO2, MeO, Br] were synthesized by the reaction of the 1,2-ethylenediamine with 2-hydroxybenzaldehyde and its derivatives (Scheme 1). Moreover, cobalt(III) Schiff base complexes were successfully synthesized in ethanol and characterized by FT-IR, 1HNMR, 13CNMR, 31PNMR and UV–Vis spectroscopy. X-ray crystallography was used to determine the coordination geometry of [Co(5-BrSalen)(PEt3)(H2O)]ClO4 compound. In a follow-up study, nanohybrid of the synthesized complexes and Montmorillonite-K10 (MMT-K10) was prepared in methanol and their properties were determined by different techniques such as SEM, TEM, TGA-DTG, EDX and XRD. The electrochemical method provides highly valuable information on the catalytic processes.

Structure of Schiff bases and their complexes
Experimental
Materials
All the chemicals and solvents were commercially available reagent and used as received without further purification. MMT-K10 clay (cation-exchange capacity, 119 meq/100 g) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Characterizations
Fourier transform infrared (FT-IR) spectra of the samples were recorded using a FT-IR JASCO-680 spectrophotometer in the range of 4000–400 cm−1 at room temperature with 16 scan times. The UV–Vis spectra were recorded on a JASCO V-570 spectrophotometer in the range of 190–900 nm. The 1H NMR, 13C NMR and 31P NMR spectra were recorded in DMSO-d6 on Bruker-500 MHz. The Bruker smart apex II-2009 CCD area detector diffractometer was used to obtain the X-ray single crystal structure of the metal complex. The X-ray diffraction (XRD) analysis was performed by high resolution X-ray diffractometer system with the X’Pert model (MRD PANalytical, Almelo, Netherlands). Scanning electron microscopy (SEM) was recorded on (Quanta FEG 650 2012, FEI, Hillsboro, OR) system. Transmission electron microscopy (TEM) was performed using Zeiss Libra (120, Carl Zeiss AG, Oberkochen, Germany) system. The thermal stability of specimens was tested using (PerkinElmer TGA7 1991, Waltham, MA, USA) thermogravimetric analyzer from ambient temperature to 900 °C at a heating rate of 20 °C/min under nitrogen gas. Cyclic voltammograms were carried out by a SAMA Research Analyzer M-500. A conventional three-electrode Ag wire and a glassy carbon were used as a reference and a working electrode, respectively. The working electrode (Metrohm 6.1204.110, with 2.0 ± 0.1 mm diameter) was polished and cleaned with 1 μm alumina prior to each scan. Tetrabutylammonium perchlorate (TBAP) was used at the supporting electrolyte. The solution was deoxygenated by Ar for 5 min. All electrochemical potentials were calibrated versus an internal Fc+/0 (E0 = 0.45 V vs. SCE) couple under the same condition [27].
Synthesis of the Schiff base ligands
The Schiff base ligands were synthesized by condensation of 1,2-ethylenediamine and benzaldehyde derivatives (1:2 mol ratio) in methanol [28].
X2Salen; FT-IR (KBr cm−1) ν max X = H (C=N)(1635), (C=C)(1577), (C–O)(1283); X = MeO (C=N)(1617), (C–C)(1492), (C–O)(1265); X = Br (C=N)(1634), (C=C)(1473), (C–O)(1276); X = NO2 (C=N)(1612), (C=C)(1540), (C–O)(1215), (NO2)(1324).
UV–Vis, λ max (nm) (Ethanol): X = H 318(11,400), 260(31,100), 216 (57,600); X = MeO 346(17,700), 258(29,700), 230(47,900); X = Br 322(7700), 247(29,500), 224(44,700); X = NO2 374(24,000), 250(25,100), 236(27,000).
Synthesis of the Schiff base metal complexes
1.0 mmol of cobalt(II)acetatetetrahydrate (0.249 g) and 1.0 mmol of triethylphosphine ((0.144 ml) were added to an ethanolic solution of X2Salen (1.0 mmol, X = H: 0.268 g, NO2 0.358 g, MeO 0.328 g and Br 0.426 g) and refluxed under inert atmosphere for 2 h. The solution was subjected by blowing air for 2 h to oxidize the Co(II) complex, and then, it was filtered. Furthermore, 1.0 mmol of sodium perchlorate (0.140 g) was added to the filtrate, and the resulting crystal was appeared after 5 days. The suitable crystals of [Co(5-BrSalen)(PEt3)(H2O) [ClO4 for X-ray were obtained from methanol after 5 days.
[Co(Salen)(PEt3)(H2O)[ClO4; FT-IR (KBR cm−1) ν max (C=N) (1625), (C=C) (1448), (C=O)(1304), (C–H)(2870–3058), (ClO4 −)(1092). UV–Vis (λ max) (nm)(Ethanol) (626)(840), (401)(7400), (320)(9200), (256)(66,000), (210)(28,100); 1H NMR (DMSO-d6, δ, ppm): 0.905–0.967 (m, 9H, CH3), 1.458–1.528 (m, 6H, CH2), 3.949 (s, 4H, CH2 bridge), 6.610–6.639 (t, 2Ar-H, 3 J = 7.5), 7.062–7.079 (d, 2Ar-H, 3 J = 7.5), 7.297–7.343 (m 4Ar-H), 8.255–8.260 (d, 2HC=N, 5 J = 2.5). 13C NMR (DMSO-d6, δ, ppm): 7.51–7.57 (d, 4 J = 3, CH3), 14.68–14.88 (d, 3 J = 10, CH2), 58.79 (CH2 bridge), 115.62, 119.52, 122.49, 134.68, 135.31 and 164.74 (C-Ar), 167.98 (CH=N). 31P NMR (DMSO-d6, δ, ppm): 40.73.
[Co(5-MeOSalen)(PEt3)(H2O)[ClO4; FT-IR(KBr cm−1) ν max (N)(1629), (C=C)(1471), (C–O)(1294), (C–H) (2834–3045), (ClO4 −)(1092); UV–Vis (λ max) (nm)(Ethanol), (654)(500), (428)(13,400), (256)(48,400), (210)(28,900); 1H NMR (DMSO-d6, δ, ppm): 0.920–0.969 (m, 9H, CH3), 1.446–1.516 (m, 6H, CH2), 3.949 (s, 4H, CH2), 6.862–6.866 (d, 2Ar-H, 4 J = 2.5), 6.997–7.053 (m, 2Ar-H), 7.297–7.343 (m, 4Ar-H), 8.181–8.210 (d, 2HC=N, 5 J = 2.5). 13C NMR (DMSO-d6, δ, ppm): 7.56–7.61 (d, 4 J = 2.5, CH3), 14.63–14.83 (d, 3 J = 10, CH2), 55.90 (O–CH3), 58.76 (CH2 bridge), 114.95, 118.22, 123.18, 124.57, 149.39, and 159.91 (C–Ar), 167.47 (CH=N). 31P NMR (DMSO-d6, δ, ppm): 39.76.
[Co(5-BrSalen)(PEt3)(H2O) [ClO4; FT-IR(KBr cm−1) ν max (C=N) (1631), (C=C) (1456), (C–O) (1302), (C–H) (2870–3070),(ClO4 −) (1088); UV–Vis (λ max) (nm) (Ethanol), (630) (500), (412)(2700), (320)(4700), (256)(67,900), (205)(27,200); 1H NMR (DMSO-d6, δ, ppm): 0.913–0.992 (m, 9H, CH3), 1.473–1.567 (m, 6H, CH2), 3.946 (s, 4H, CH2), 7.004–7.022 (d, 2Ar-H, 3 J = 10), 7.397–7.420 (dd, 2Ar-H, 3 J = 10, 4 J = 2.5), 7.550–7.555 (d 2Ar-H, 4 J = 2.5), 8.233–8.239 (d, 2HC=N, 5 J = 2.5). 13C NMR (DMSO-d6, δ, ppm): 7.56–7.61 (d, 4 J = 2.5, CH3), 14.91–15.11 (d, 3 J = 10, CH2), 59.06 (CH2 bridge), 105.71, 121.17, 124.68, 135.97, 137.61 and 163.73 (C–Ar), 167.53 (CH=N). 31P NMR (DMSO-d6, δ, ppm): 42.71.
[Co(5-NO2Salen)(PEt3)(H2O)[ClO4;FT-IR(KBr cm−) ν max (C=C) (1466), (C=N) (1633), (C–H) (2885–3110),(ClO4) (1105),(N–O) (1314); UV–Vis (λ max) (nm)(Ethanol), (635)(900) (366)(31,600), (252)(43,900), (212)(26,500); 1H NMR (DMSO-d6, δ, ppm): 0.934–0.996(m, 9H, CH3), 1.554–1.594 (m, 6H, CH2), 4.014 (s, 4H, CH2), 7.170–7.188 (d, 2Ar-H, 3 J = 9), 8.146–8.170 (m, 2Ar-H), 8.494–8.506 (m 2Ar-H, and 2HC=N). 13C NMR (DMSO-d6, δ, ppm): 7.60–7.66 (d, 3 J = 3, CH3), 15.29–15.49 (d, 3 J = 10, CH2), 59.32 (CH2 bridge), 119.00, 123.05, 129.58, 132.42, 136.79 and 169.01 (C–Ar), 170.24 (CH=N). 31P NMR (DMSO-d6, δ, ppm): 46.60.
Synthesis of intercalation compounds
An appropriate amount of MMT-K10 (0.75 g) was dispersed to methanolic solution containing [Co(Salen)(PEt3)(H2O)[ClO4, [Co(5-NO2Salen)(PEt3)(H2O)[and [Co(Salen)(PBu3)(H2O)[ClO4 (about 0.08 g) complexes. The reaction suspension was further refluxed for 24 h. The green precipitates were filtered and washed several times with methanol and acetone.
K10-[Co(Salen)(PEt3)(H2O)[ClO4: FT-IR (KBr cm−1) ν max; 3200–3600 (H2O), 1631 (C=N), 1048 (Si–O), 523 (Mg–O), 464 (Al–O) and 1088–1105 cm−1 (Cl–O).
K10-[Co(NO2Salen)(PEt3)(H2O)[ClO4: FT-IR (KBr cm−1) ν max; 3200–3600 (H2O), 1630 (C=N), 1048 (Si–O), 522 (Mg–O), 464 (Al–O) and 1088–1105 cm−1 (Cl–O).
K10-[Co(HSalen)(PBu3)(H2O)[ClO4: FT-IR (KBr cm−1) ν max; 3200–3600 (H2O), 1631 (C=N), 1047 (Si–O), 522 (Mg–O), 464 (Al–O) and 1088–1105 cm−1 (Cl–O).
Electrochemistry
The electrochemical properties of the complexes were studied by cyclic voltammetry at ambient temperature. The voltammograms were obtained from approximately 10−3 M DMF solutions with a working pt, pt auxiliary and Ag/AgCl reference electrodes in the range of − 2 to 0.0 V. The tetra-n-butylammonium perchlorate (TBAP) (0.1 M) and ferrocene were used as supporting electrolyte and internal standard to reference the data versus Fc+/0 couple, respectively [29].
Determination refinement of [Co(5-BrSalen)(PEt3)(H2O)[ClO4 crystal structure
For [Co(5-BrSalen)(PEt3)(H2O)[ClO4 complex, green crystal with a dimension of 0.50 × 0.18 × 0.08 mm was mounted on a glass fiber for the data collection. The least squares refinement of diffraction data from 9940 unique reflections was used to obtain the cell constants and an orientation matrix. Data were recorded at 297 K to a maximum 2θ value of 50° in a series of ω scans in 1° oscillations, and the Stoe X-AREA software package was used to integrate the data. The numerical absorption coefficient (μ) for Mo-Kα radiation is 3.46 mm−1.
A numerical absorption correction was performed using X-RED and X-SHAPE software [30], and the data were reformed for Lorentz and polarizing effects. The structure was solved by direct methods [31] and subsequent difference Fourier maps and then refined on F 2 by a full-matrix least squares procedure using anisotropic displacement parameters [32]. The hydrogen atoms were in a difference Fourier map and consequently refined isotropically. Atomic factors were from international tables for X-ray Crystallography. The X-STEP32 crystallographic software package was used to perform all the refinements [33].
Computational details
The geometries of the molecules in both the ethanol and gas phase have been optimized with Gaussian 09 program package [34] at B3LYP/6-31 + G(d) level of theory. To modeling the solvent in the calculations, the PCM (polarized continuum model) has been used. The IR spectra of the molecules have been calculated at the same level of theory. UV–Vis spectra calculations were obtained from time-dependent density functional theory (TD-DFT) with the same functional and basis.
Result and discussion
FT-IR characteristics
The FT-IR spectra of the Schiff base ligands and cobalt(III) complexes exhibit several bands in the 400–4000 cm−1 region (experimental section). The azomethine vibration of the Schiff base ligands was appeared in the range of 1617–1635 cm−1. In the formed complexes, the bond formation between the metal and the azomethine caused the C–N bond stretching shifted to the lower frequencies (bathochromic shift) relative to the free Schiff bases [35]. The peak in the range of 2870–3110 cm−1 is associated with C–H stretching of PEt3 coordinated ligand. However, the peak in the range of 1088–1105 cm−1 is due to the stretching vibrations of ClO4 − counter ion.
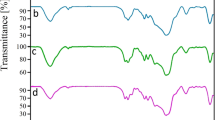
MMT-K10 showed a broad peak in the range of 3200–3600 cm−1 which is assigned to the interlayer and physically absorbed water molecules on the surfaces. OH bending showed a weak band at 1645 cm−1, and the wide Si–O stretching peak is appeared in the range of 900–1300 cm−1. The peaks at 463 and 529 cm−1 are related to Mg–O and Al–O, respectively [36]. The FT-IR spectra of the intercalation compounds are illustrated in Fig. 1.

FT-IR spectra of the MMT-K10 and complexes
Electronic spectroscopy
The full-range UV–Vis spectral data of the ligands and synthesized complexes were shown in experimental section. All the peaks below 400 nm are involved to π → π* transition which is related to the aromatic ring in the Schiff base ligands. However, the peak at 453 nm involves to π → π* transition assigned to azomethine group. This peak was shifted to higher wavelengths (490 nm) with the charge transfer transition [17, 37,38,39,40,41]. The cobalt(III) complexes revealed a d–d transition around 650–700 nm [18, 19].
1H NMR, 13C NMR and 31P NMR spectroscopy
The 1HNMR data of the synthesized cobalt(III) complexes were showed in the experimental part. The hydrogen of the azomethine was seen in the range of 8.181–8.506 ppm as a doublet peak in the different complexes. The hydrogens of the aromatic rings (6.610–7.555 ppm) were doublet and triplet (doublet of doublet) peaks. Some of these hydrogens on NO2Salen were appeared in low field and mixed with azomethine hydrogens. The bridge hydrogens were appeared in the range 3.946–4.162 ppm. The hydrogens of PEt3 were appeared in the range 1.446–1.594 and 0.905–0.996 ppm for CH2 and CH3 groups, respectively. These hydrogens split with phosphine so were seen as multiplet peaks. The 1H NMR spectrum of [Co(5-BrSalen)(PEt3)(H2O)]ClO4 complex showed these hydrogens clearly (Fig. 2).

1H NMR of [Co(5-BrSalen)(PEt3)(H2O)]ClO4 complex taken into DMSO-d6
Furthermore, 13C NMR was used to confirm the structures of the synthesized complexes. The numbers of peaks were equal to the number of carbons in the complexes. The imine carbon was appeared in the range of 167.53–169.01 ppm. The six-different lines, belong to the aromatic carbons, were seen in the range of 105.62–170.24 ppm. The carbons of CH2 bridged appeared in the range of 57.79–59.32 ppm. The carbons of PEt3 were seen in the range 7.51–15.49 ppm as two doublet peaks.
The 31P NMR of complexes was also recorded in DMSOd6 solvent. The peak of phosphorus in the PEt3 was appeared at 39.76, 40.73, 42.71 and 46.60 ppm in the MeOSalen, Salen, BrSalen and NO2Salen complexes, respectively. Based on these results, the increase in the electron-withdrawing power of the functional groups on the Schiff base ligands caused the phosphorus peak displaced to the lower magnetic field. Also, there is a good correlation between the Hammett parameter due to the functional groups on the Schiff bases and the place of 31P NMR peaks (Fig. 3). This trend shows that in a complex with weak Schiff base ligand, like [Co(NO2Salen)(PEt3)(H2O)]+, because of withdrawing functional groups, the electron density on the phosphorous is lower than a complex containing a Schiff base with donor functional groups, like MeOSalen.

Correlation between the phosphorous chemical shift values for complexes and the respective σ pa substituent
Electrochemistry
The electrochemical activities of the synthesized complexes were studied at ambient temperature using DMF solvent with 0.1 M tetrabutylammonium perchlorate (TBAP, as supporting electrolyte) at a glassy carbon working electrode in the potential range from 0.0 to − 2.0 V. Ferrocene (Fc) was employed as the internal standard, and Fc+/0 couple was referenced for all redox potentials.
A typical cyclic voltammogram of [Co(XSalen)(PEt3)(H2O)[+ complex is shown in Fig. 4. The first peak reduction observed at a bout − 0.44 V is related to reaction 1:
In this reaction, electron added to the dz2 orbital and the product losses its H2O ligand [19, 20, 42]. The second peak is related to reaction 2:
Based on the results, all the redox processes in studied complexes have both two electron transfer reactions. Table 1 shows the oxidation potentials for all the different complexes. The formal potentials E 1/2 of the redox couple were calculated based on the average of anodic (E pa) and cathodic (E pc) potentials peaks. The cyclic voltammetry was used for a series of cobalt Schiff base complexes to investigate the possible effect of functional groups of Schiff base ligands on the oxidation potentials of [Co(5-XSalen)(PEt3)]ClO4. Based on the results, the E pa varies due to the electronic effects of the substituents at position five. Thus, E pa becomes more positive per the sequence MeO < H < Br < NO2. Comparable results have been reported for cobalt(III), vanadyl(IV), copper(II) and nickel(II) systems, which assuming that the strong electron-withdrawing effects can stabilize the lower oxidation state, while electron-donating groups have a reverse mechanism [43,44,45]. Hammett-type relationship was found between the appropriate para-substituent parameters and E(II/I)1/2 values with variation of electrode potential as a function of electron-withdrawing ability due to substituent at position 5 (Fig. 5).

Cyclic voltammogram of [Co(Salen)(PEt3)(H2O)]ClO4 complex in DMF solvent, scan rate = 100mv/sec

Correlation between the E pa values for complexes and the respective σ pa substituent
Description of the [Co(5-BrSalen)(PEt3)(H2O)]ClO4
The X-ray diffraction was used to determine the structure of the [Co(5-BrSalen)(PEt3)(H2O)[ClO4 complex. The complex was crystallized in the monoclinic space group P2 1 / C prior to measurement. A summary of crystal data, experimental details and refinement result are given in Table S1. Moreover, angles and bond lengths of [Co(5-BrSalen)(PEt3)(H2O)[ClO4 complex are listed in Table S2. Furthermore, the asymmetric unit of [Co(5-BrSalen)(PEt3)(H2O)[ClO4 complex is shown in Fig. 6.

a The labeled diagram of [Co(5-BrSalen)(PEt3)(H2O)]ClO4 showing 65% probability thermal ellipsoids, b the optimized structures of [Co(5-BrSalen)(PEt3)(H2O)]+ calculated at the M062X/LANL2DZ/6-31G(d)
In this complex, the Salen Schiff base ligand has the N2O2 coordination in the equatorial pane and the central cobalt atom is coordinated in a distorted octahedral geometry. The P–Co–N/O and O1w–Co–N/O angles are distributed from 97.43 (17)° to 87.83 (13)° and from 88.83(19)° to 87.91(17)°, respectively. The distances of Co–N1, Co–N2, Co–O1 and Co–O2 appeared in the range of 1.876 (5), 1.880 (5), 1.883(4) and 1.884(4) Å, respectively. The results are comparable with N2O2-Salen cobalt complexes [46,47,48]. The Co-p bond distance of apical position appeared at 2.214(19). According to the higher transinfluence of the P1 atom with regard to the N1 and N2 atoms, the Co-O distances for Schiff base (1.883(4) 1.884(4) Å) were smaller than the Co-Ow distance 2.074 (4) Å.
The network of the complex is extended due to the intermolecular interaction (hydrogen bonding) in molecules which lead to aggregate the supramolecular structure (Fig. 7).

A view of the extended network of complex [Co(5-BrSalen)(PEt3)(H2O)]ClO4 showing 20% probability thermal ellipsoids with the intermolecular hydrogen bonds (dashed lines)
Based on the regular distortion, all the angles surrounded by cobalt center deviate significantly from 90°. The ligand–cobalt–ligand bonds in the equatorial plane consist of two larger and two smaller angles than 90°. The obtained data are similar and comparable to those N2O2–Salen cobalt complexes [49]. The summation of angles around 360° indicates the cobalt atom exists in a square planer environment of N2O2 atoms.
Theoretical section
Table S2 shows the experimental and the calculated results for some important bond lengths and bond angles in the [Co(5-BrSalen)(PEt3)(H2O)[+ complex. Its optimized geometry is shown in Fig. 6b. As indicated, there is an excellent agreement between the calculated and experimental data, which confirms the accuracy of the applied theoretical method. So, it seems for the other molecules, in the absence of X-ray data, the theoretical results are accurate enough to determine their structure. In addition, some important geometrical parameters of other studied complexes are gathered in Table S3.
Figure 8 compares experimental and calculated IR spectra of [Co(5-BrSalen)(PEt3)(H2O)[+ complex. Some characteristic peaks derived from calculated results and the experimental data are shown. The harmonic DFT frequencies were scaled with 0.959 scaling factor [50]. Again, a good agreement between calculation and experiment is observed. For instance, the calculated C=N vibrational frequency is observed at 1609 cm−1 for [Co(5-BrSalen)(PEt3)(H2O)[+ complex that is closed to the experimental value (1630 cm−1). The C–H vibrational frequencies of PEt3 coordinated to cobalt(III) are seen in the range of 2938–3016 cm−1. The calculated stretching frequencies of coordinated water are seen in the range of 3490 and 3611 cm−1 as weak and sharp peaks.

Experimental and theoretical (red spectrum) IR spectra of [Co(5-BrSalen)(PEt3)(H2O)]ClO4
Theoretical and experimental UV–Vis spectra of the [Co(5-BrSalen)(PEt3)(H2O)[+ compound are depicted in Fig. 9. All the results have been recorded in ethanol solvent. Some important absorptions are collected in Table S3. As one can see from the feature of figures and the absorption bands, there is a good agreement between the two sources of data.

Experimental (black, b) and theoretical (red spectrum, a) UV–visible spectra of [Co(5-BrSalen)(PEt3)(H2O)]ClO4
The first absorption peak (664 nm) is due to excitation from the HOMO (π + d) to the LUMO (d + σ*) orbitals. The second line (468 nm) is due to the promotion of electron from HOMO − 4 to LUMO + 3. The most intense absorption peak at about 385 nm (in the calculated spectrum) contains several bands. The most intense band in this region is derived from the excitation of electron from HOMO to LUMO + 2 orbitals. Finally, the absorption band at about 289 nm is an excitation from HOMO − 3 to LUMO orbitals.
X-ray diffraction
To investigate the electronic and steric effects of cobalt Schiff base complexes incorporated in to the clay, our studies were extended to the interaction of cobalt Schiff base complexes with Montmorillonite-K10 nanoclay [24, 25] and tri types of complexes, involves [Co(Salen)(PEt3)(H2O)]ClO4, [Co(Salen)(PBu3)(H2O)]ClO4 and [Co(5-NO2Salen)(PEt3)(H2O)]ClO4 were interacted with Montmorillonite-K10 nanoclay.
Figure 10 demonstrates the XRD fingerprints of the K10-CoPEt3 and K10-CoPBu3. The basal spacing (1.006 nm) of pure clay (K10) was calculated from the peak position at 2θ = 8.78° using Bragg’s equation [24]. After interaction of MMT with the complexes, the basal spacing peak of the K10-CoPEt3 and K10-CoPBu3 appeared at 2θ = 8.81° (d = 1.00316 A) and at 2θ = 8.80o (d = 1.00389 A), respectively, with a little different relative to the Montmorillonite-K10. According to these results, the intercalation reaction dose not occurred partially and the complexes absorbed on the surface of the clay.

XRD pattern of the MMT-K10 with complex [Co(Salen)(PEt3)(H2O)]ClO4 and [Co(Salen)(PBu3)(H2O)]ClO4
SEM and TEM analysis
A surface of K10-[Co(Salen)(PEt3)(H2O)]ClO4 was examined by SEM technique and seems smooth with a layer structures (Fig. 11). Furthermore, the TEM microimages of K10-[Co(Salen)(PEt3)(H2O)]ClO4 nanohybrid are presented in Fig. 12. Based on the TEM microimages obtained, the thickness of the organoclay layers is in the rage of nanocompounds.

SEM image of the K10-[Co(Salen)(PEt3)(H2O)]ClO4

TEM image of the K10-[Co(Salen)(PEt3)(H2O)]ClO4
Thermal properties
The K10-[Co(Salen)(PEt3)(H2O)]ClO4 decomposes into two steps (Figures S1 and S2); the mass loss (7.5%) at ambient temperature to 207 °C is due to the moisture and physically adsorbed water. The second step of decomposition (with 11.5% mass loss) is related to the bonded water and decomposition of complex. The K10-[Co(NO2Salen)(PEt3)(H2O)]ClO4 decomposes into two steps, i.e., the weight loss (10%) at ambient temperature to 380 °C is due to the moisture and the absorbed complexes. The second decomposition step (8.5%) is related to the bonded water and absorbed complex on the surface of clay.
EDX
The EDX pattern of the MMT-K10 [Co(Salen)(PEt3)(H2O)]ClO4 is shown in Fig. 13. In the analysis, existence ingredient of Co, N and P characterized that complex interacted with nanoclay.

EDX of MMT-K10 [Co(Salen)(PEt3)(H2O)]ClO4 composite
Supplementary material
CCDC No. 1472361 contains the supplementary crystallographic data for [Co(5-BrSalen)(PEt3)(H2O)]ClO4. These data can be obtained at www.ccdc.cam.ac.uk/deposit (or from the Cambridge Crystallographic Data Center 12, Union Road Cambridge CB2 1EZ, UK; Fax: (internet) +44-1223/336-033; E. mail: deposit@ccdc.cam.ac.uk).
References
M.M. Bernardo, M.J. Heeg, R.R. Schroeder, L.A. Ochrymowycz, D.B. Rorabacher, Inorg. Chem. 191, 31 (1992)
P.H. Hang, J.G. Keck, E.J. Lien, M. Mc Lai, J. Med. Chem. 608, 33 (1990)
A.E. Tai, E.J. Lien, M. Mc Lai, T.A. Khwaja, J. Med. Chem. 236, 27 (1984)
M. Dostani, A.H. Kianfar, W.A. Kamil Mahmood, M. Dinari, H. Farrokhpour, F. Abyar, M.H. Azarian, Spectrochim. Acta, Part A 180, 144 (2017)
M. Sedighipoor, A.H. Kianfar, W.A.K. Mahmood, M.H. Azarian, Polyhedron 129, 1 (2017)
L. Canali, D.C. Sherrigton, Chem. Soc. Rev. 28, 85 (1998)
A.A. Isse, A. Gennaro, E. Vianello, J. Electroanal. Chem. 444, 241 (1998)
D. Pletcher, H. Thompson, J. Electronal. Chem. 464, 168 (1999)
T. Okada, K. Katou, T. Hirose, M. Yuasa, I. Sekine, J. Electrochem. Soc. 146, 2562 (1999)
A.H. Kianfar, M. Sedighipoor, G. Mohammadnezhad, H. Görls, W. Plass, M. Roushani, J. Iran. Chem. Soc. 14, 313 (2017)
R.D. Jones, D.A. Summerville, F. Basalo, Chem. Rev. 79, 139 (1979)
S. Bhunia, S. Koner, Polyhedron 30, 1857 (2011)
H. Dugas, C. Penney, Bioorganic Chemistry (Springer, New York, 1981), p. 435
D. Ramakrishna, B. Ramachandra Bhat, R. Karvembu, Catal. Commun. 11, 498 (2010)
M.M. Tamizh, K. Mereiter, K. Kirchner, R. Karvembu, J. Organomet. Chem. 700, 194 (2012)
S.M. Polson, R. Cini, C. Pifferi, L.G. Marzilli, Inorg. Chem. 36, 314 (1997)
M. Asadi, A.H. Sarvestani, Can. J. Chem. 79, 1360 (2001)
M. Asadi, A.H. Sarvestani, B. Hemateenajad, J. Chem. Res. 1, 520 (2002)
A.H. Sarvestani, A. Salimi, S. Mohebbi, R. Hallaj, J. Chem. Res. 3, 190 (2005)
A.H. Sarvestani, S. Mohebbi, J. Chem. Res. 4, 257 (2006)
A.H. Kianfar, S. Zargari, J. Cood. Chem. 61, 341 (2008)
M. Asadi, M.B. Ahmadi, Kh Mohammadi, Z. Asadi, A.H. Sarvestani, J. Chem. Thermodyn. 36, 141 (2004)
M. Asadi, A.H. Kianfar, S. Torabi, K. Mohammadi, J. Chem. Thermodyn. 40, 523 (2008)
A.H. Kianfar, W.A. Kamil Mahmood, M. Dinari, M.H. Azarian, F.Z. Khafri, Spectrochim. Acta, Part A 127, 422 (2014)
A.H. Kianfar, W.A.K. Mahmood, M. Dinari, H. Farrokhpuor, M. Enteshari, M.H. Azarian, J. Spectrochim, Acta Part A 136, 1582 (2015)
K. Karami, S. Hashemi, M. Dinari, Appl. Organomet. Chem. 31, 3672 (2017)
A.H. Sarvestani, M. Asadi, M. Abbasi, J. Chem. Res. 1, 56 (2007)
M. Asadi, A.H. Sarvestani, Z. Asadi, M. Setoodehkhah, Synth. React. Inorg. Metal Org. Nano Metal 35, 639 (2005)
M. Asadi, M. Setoodekhah, A.H. Kianfar, J. Iran. Chem. Soc. 7, 38 (2010)
N.G. Connelly, W.E. Geiger, Chem. Rev. 96, 877 (1996)
J. Kjeld, C. van Bommel, W. Verboom, H. Kooijman, A.L. Sprk, N.D. Reinhoudt, Inorg. Chem. 37, 4197 (1998)
G. Dacarro, L. Cucca, P. Grisol, P. Pallavicini, M. Patrini, A. Taglietti, Dalton Trans. 41, 2456 (2012)
G.M Sheldrick, SHELX97. Program for crystal structure solution, University of Gottingen, Germany, 1997
G.M. Sheldrick, Acta Cryst. A 64, 112 (2008)
International Tables Foe X-ray Crystallography, Vol. C, Kluwer Academic Publisher, Doordrecht, The Netherlands 1995
Bruker. SADABS. Bruker AXS Inc., Madison, Wisconsin, USA 2005
Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT
A.H. Kianfar, V. Sobhani, M. Dostani, M. Shamsipur, M. Roushani, Inorg. Chim. Acta 355, 108 (2011)
N.S. Biradar, V.H. Kulkarni, J. Inorg. Nucl. Chem. 8, 2451 (1971)
N.S. Biradar, G.V. Karajagi, T.M. Aminabhavi, Inorg. Chem. Acta 82, 211 (1984)
J.A. Bertrand, P.G. Eller, Inorg. Chem. 4, 927 (1974)
E. Ochiai, K. Long, C.R. Sperati, D.H. Busch, J. Am. Chem. Soc. 91, 3201 (1969)
D.F. Shriver, P.W. Atkins, Inorganic Chemistry (Oxford University Press, Oxford, 1999), p. 240
Y.L. Zhang, W.J. Ruan, X.J. Zhao, H.G. Wang, Z.A. Zhu, Polyhedron 22, 1535 (2003)
A. Bottcher, T. Takeuchi, I. Hardcastle, T.J. Mead, H.B. Gray, D. Cwikel, M. Kapon, Z. Dori, Inorg. Chem. 36, 2498 (1997)
A.H. Sarvastani, S. Mohebbi, J. Iran. Chem. Soc. 4, 215 (2007)
E.G. Jager, K. Schmann, H. Gorls, Inorg. Chem. Acta 255, 295 (1997)
S. Zlezzi, E. Spodine, A. Decinti, Polyhedron 21, 55 (2002)
J. Welby, L.N. Rusere, J.M. Tanski, L.A. Tyler, Inorg. Chem. Acta 362, 1405 (2009)
K.K. Irikura, R.D. Johnson III, R.N. Kacker, J. Phys. Chem. A 109(37), 8430 (2005)
Acknowledgements
We wish to express our gratitude to the Research Affairs Division Isfahan University of Technology (IUT), Isfahan, for partial financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kianfar, A.H., Tavanapour, S., Eskandari, K. et al. Experimental and theoretical structural determination, spectroscopy and electrochemistry of cobalt (III) Schiff base complexes: immobilization of complexes onto Montmorillonite-K10 nanoclay. J IRAN CHEM SOC 15, 369–380 (2018). https://doi.org/10.1007/s13738-017-1238-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-017-1238-2