
Abstract
Edible oils are used in the preparation of foods as a part of their recipe or for frying. So to ensure of food safety, checking the quality of the oils before and after usage is an important subject in food control laboratories. In this study, edible oils from four different sources (canola, corn, sunflower and frying) were heated for 36 h at 170 °C and sampling was done every 6 h. The free fatty acid, peroxide value and the content of some fatty acids (C16:0, C18:0, C18:1, C18:2, C18:3) of the oil samples were determined by standard methods. Then, the ATR-FTIR spectra of the samples were collected. The partial least squares (PLS) regression combined with genetic algorithm was performed on the spectroscopic data to obtain the appropriate predictive models for the simultaneous estimation of acid value, peroxide value and the percentage of five kinds of fatty acids. The effect of some preprocessing methods on these models was also investigated. Preprocessing of data by orthogonal signal correction (OSC) resulted in the best predictive models for all oil properties. The correlation coefficients of calibration set (>0.99) and validation set (>0.86 and in most case >0.94) of the OSC–PLS model suggested suitable predictive modeling for all studied parameters in the oil samples. This method could be suggested as a rapid, economical and environmental friendly technique for simultaneous determination of seven noted parameters in the edible oils.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Edible oils are common in world for food making as a part of their recipe or for frying of food stuffs. Some chemical reactions such as hydrolysis, oxidation and polymerization occur on the oil during heating. The generation of free fatty acids, primary and secondary oxidation products, monoglyceride, diglyceride and some other compounds are the result of such reactions [1]. As the consequence of these reactions, the taste, flavor and color of the oils are changed and simultaneously the safety of oil is affected and may cause side effects on human health [2, 3].
The security of food is related to the safety of the oil used in its preparation [4]. So the quality control of oils is an important subject in the food laboratories. Various chemical, physicochemical and physical parameters have been used to evaluate the quality of oils and being used as standards to ensure their safety [5]. Quality assurance methods of oil which are commonly used are determination of free fatty acids (FFA), the peroxide value (PV) and the percentage of fatty acids (FAs) of the oils [6].
The FFA (as a scale for the acidity of oil) is an explanatory parameter for evaluation of the level of hydrolysis of oil. In other words, the formation of FFA and glycerol residues is the result of hydrolysis reaction. The FFA usually can be determined by acid/base titration.
The level of peroxides/hydroperoxides formed at initial process of oil oxidation is determined by iodometric titration and is known as peroxide value [5]. In addition to limitation of reproducibility and sensitivity in these titrimetric methods, the involving health hazardous solvents and chemicals are the other problems along with the noted procedure [6].
On the other hand, the common method for determination of the percentage of FAs in oils is gas chromatography (GC), which is a time-consuming method in this subject, because it requires a sample preparation step before injection to GC apparatus which includes the hydrolysis of triglycerides to form FFAs and conversion of them into methyl esters [5, 7].
It should be noted that multivariate analysis adds some significant advantages for analysis of complex mixtures [8–11]. Combination of multivariate methods with ATR-FTIR spectroscopy enable one to perform simultaneous analysis and determination of some parameters in the desired samples with no or minimum sample preparation and with saving a lot of time as well [6, 12].
FTIR spectroscopy combined with multivariate chemometrics analysis has been applied for determination of some parameters in edible oils such as FFA [6], PV [6, 12], and fatty acid percentage of oleic acid (C18:1), linoleic acid (C18:2) [4, 12], and some other parameters in edible oils, but the focus of the current research is more complete view to determine seven important parameters including FFA content or acid value (AV), PV and the percentage of five kinds of FAs such as palmitic acid (C16:0), stearic acid (C18:0), oleic acid (C18:1), linoleic acid (C18:2) and linolenic acid (C18:3) simultaneously in four type of edible oils (canola, corn, frying and sunflower oil) during 36-h heating at 170 °C. In other words, the variation of oil’s composition during heating process is the main objective of this study.
The proposed method can be considered as an alternative rapid technique for identification of oil type based on the determination of the composition of FAs and also for evaluation of oils’ quality applied in food preparation by estimation of their FFA content and peroxide value.
Experimental
Samples
Fourteen oil samples with various brands from four types of edible oils, including Canola (4 samples), Corn (4 samples), Sun flower (3 samples) and frying, which was included a mixture of palm, sunflower and soybean or cottonseed oil (3 samples), were purchased from local markets at Shiraz, Iran.
Heating procedure of oils
All of the oil samples were heated at 170 °C for 36 h by a thermostatic heater, and sampling was done every 6 h from each of the heated oils. So, in addition to samples collected before heating, six other samples were collected from each oil. These samples were stored in sealed vials at the temperature of 4–6 °C in dark until the analysis.
Fatty acids determination
Gas chromatography-mass spectroscopy (GC–MS) was utilized for identification of the FAs composition in each oil type and %fatty acids was determined using gas chromatography-flame ionization detector (GC–FID) according to ISO 5508: 1990 [13]. For this purpose, GC analysis of the fatty acids methyl ester was performed and the quantification was based on the peak area of each component in the chromatogram.
For conversion of FAs to their methyl ester according to ISO 5509: 1978 [14], 0.5 mL methanolic KOH (1 M) was added to the methanolic solution of oil (0.1 g/40 mL) and the methyl esterification of FAs was performed under the reflux condition for 10 min; afterward the decantation was accomplished by two stages addition of 40 mL water to the mixture of the reaction products in 40 mL n-heptane.
GC–FID analysis of fatty acids methyl ester was carried out using an Agilent 6890 N chromatograph (Agilent Technologies, USA) with a DBWAX capillary column (polyethylene glycol, 30 m × 0.32 mm; 0.25 µm film thickness). The oven temperature increased from 180 to 220 °C at a rate of 4 °C/min for 10 min, and the injector and detector temperatures were 210 and 230 °C, respectively. Helium was used as the carrier gas with a flow rate of 0.75 mL/min and a split ratio of 1:60.
GC–MS analysis of the obtained fatty acids methyl ester performed using an Agilent 7890A chromatograph, coupled with an Agilent 5975C mass spectrometer (Agilent Technologies, USA), operating at 70 eV ionization energy, 0.5 s/scan and the mass range: 30–300 amu, equipped with a DBWAX capillary column (polyethylene glycol, 30 m × 0.32 mm; 0.25 µm film thickness) programmed as above with helium as the carrier gas with the flow rate of 0.75 mL/min and a split ratio of 1:60. Chem Station Software was utilized to handle mass spectra and gas chromatograms.
Free fatty acid analysis
Free fatty acids content or acid value was determined according to ISO 660:2009 [15] by the acid–base titration of a solution of oil in hot neutral ethanol (3g/10 mL) with 0.1 M KOH using ethanolic phenolphthalein solution as the indicator.
Peroxide value
Peroxide value is defined as milli-equivalents of oxygen kg−1 of oil (meq O2/kg of oil) and was determined according to ISO 3960:2007 [16] by dissolving the oil samples in the mixture of isooctane and glacial acetic acid (40:60) and allow to react with freshly saturated solution of potassium iodide (KI) for 60 s. Free iodine (I2) was determined by titration of the desired mixture with standard sodium thiosulfate (Na2S2O3; 0.01 M)) in presence of starch solution (1 g/100 mL) as the indicator.
FT-IR spectroscopy
IR spectra of the oil samples were recorded in absorption mode, with a PerkinElmer RX1 FT-IR spectrometer equipped with an attenuated total reflectance (ATR) cell (ZnSe Flat plate, 01873, PerkinElmer). The spectra were collected in the range 4000–550 cm−1 at room temperature. A background spectrum was recorded at the beginning of each sample measurement and was used for background correction.
Data analysis
PLS and GA–PLS regression and other calculation were performed using PLS-toolbox in MATLAB environment (version 7.12.0.635 Mathwork, Inc., http://www.mathworks.com, USA).
Data sets
For each oil sample, the set of seven ATR-FTIR runs for each oil type (one sample before heating procedure and six samples which were collected every 6 h during 36 h of heating) gave a matrix, sized 7 × 3451. After appending of these matrices of all tested oil samples (14 oil samples of four oils’ type) one on top of each other (column wise augmentation), another matrix formed with the size of 98 × 3451. The rows of this matrix contain oils’ spectra of the collected samples during the heating procedure and the columns include the wavenumber variables. By random splitting of total 98 rows, 78 samples were utilized as the training set for model development and remaining 20 samples were reserved as the test set to evaluate the ultimate performances of the developed models. So, two new matrices formed the training or calibration data set (78 × 3451) and validation or test data set (20 × 3451).
Computational details
The acid value, peroxide value and fatty acids composition (C16:0, C18:0, C18:1, C18:2 and C18:3) of the oil samples which was heated at 170 °C for a 36-h period, were estimated using partial least square (PLS) and genetic algoritm–PLS (GA–PLS) as two well-known chemometrics treatments on the ATR–FTIR spectra of the oil samples.
The details of the theory of these well-known multivariate methods could be found elsewhere in literature [17–19] and only brifly is disscused here. Partial least squares regression (PLSR) can be considered as the developed generalization of multiple linear regression (MLR). The superiority of PLS is its ability to analyze data with strongly collinear and noisy X-variables (independent variables), and also simultaneously model several Y-variables (dependent variables).
In PLS, the X and Y matrix is decomposed in a manner similar to principal component analysis, and generate a matrix of scores (T and U), and loadings or factors (P and Q), and the residuals (E and F) for X and Y matrix, respectively. These decompositions are shown in the following equations:
Modeling of all the constituents in X and Y is the goal of PLS, so the residuals for the X and Y are approximately equal to zero. The use of information from X and Y simultaneously makes PLS more complex but more powerful than principle component regression (PCR) [20, 21].
Genetic algorithm (GA) and some kind of supervised learning multivariate calibration methods like PLS would provide a good combination for variable selection strategy. In GA, a population of n subsets which contain a random combination of variables is created. In genetic terms, each variable is called a gene and a set of variables is called a chromosome. Evaluation of the cost function for each subset is the next step in GA. Then, using techniques based on evolution and biological genetics, a new population is created. After passing the reproduction step (crossover and mutation) is the time for checking the algorithm to reach the stopping criterion. For example, this may be happened when a certain cost function response is met, a defined number of generations have passed, or the chromosomes have converged to alike configuration [22, 23].
Results and discussion
The analysis of FAs composition, AV and PV
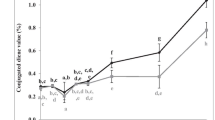
The mean of %fatty acids (C16:0, C18:1, C18:2, C18:3) in various types of studied oils which were determined by GC, is shown in Fig. 1.

Mean of %fatty acids content (C16:0, C18:0, C18:1, C18:2, C18:3), in various type of studied oils
As it is clear from fatty acids composition in Fig. 1, the studied oils have the maximum abundance of C18:1, C18:2, and contain small amounts of C18:3 and C18:0. In comparison of four types of studied oils, %saturated fatty acids (C16:0 + C18:0) in frying oil and the abundance of C18:3 in canola oil is more than the others.
To monitor the amount of PV and AV, the average of these parameters was calculated for three utilized samples (brands) of sunflower and frying oils and four utilized samples (brands) of corn and canola oils. The changing in mean peroxide value and acid value of various oil samples during 36-h heating at 170 °C is shown in Fig. 2. As it could be seen for both parameters (PV and AV), the change of profile during the time is totally ascending; however, regarding to PV (Fig. 2a) cross-sectional increasing and decreasing can be observed in its profile during the heating procedure. This increase–decrease in Fig. 2a can be due to the peroxide/hydroperoxides which are intermediate species. These intermediates are unstable species that can react very quickly.

Changing in a peroxide value and b acid value, at various tested oils (averaged for different brands in each kind of oil) during 36-h heating procedure
It was found that the AV of all of the studied oils increased slowly within 24 h of heating and after this time-point, the AV increased significantly (Fig. 2b).
To have an overall view on the variation of the FAs percentage, the amount of %fatty acids were averaged for different samples in each kind of oil and these averaged values were plotted against experimental heating time. The changing in mean %fatty acids of various studied oil samples during 36 h is shown in Fig. 3. The changing profile of averaged C16:0, C18:0, C18:1, C18:2 and C18:3 are represented in Fig. 3a–e, respectively.

Changing in %fatty acid at various tested oils (averaged for different brands in each kind of oil) during 36-h heating procedure; a C16:0, b C18:0, c C18:1, d C18:2, e C18:3
As it can be also seen in Fig. 3, the changes of mean %fatty acid profile for C16:0, C18:0 and C18:1 is totally ascending (however, C18:1 in frying oil is almost steady), and it is descending for C18:2, C18:3 during heating time.
The analysis of FTIR data
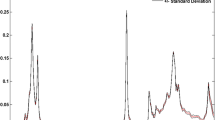
The spectra of one of the studied corn oil before and after the heating procedure as an example of recorded spectra of different oil samples are shown in Fig. 4. As it is clear from this figure, in spite of the variation in the chemical composition of samples before and after heating, their IR spectra were very similar. The spectra display characteristic bands of aliphatic hydrocarbons generated from stretching, bending and rocking vibrations at 3000–2800, 1465–1377 and 720 cm−1, respectively. C–H stretching and C–H out of plan bending of olefins at 3008 and 970 cm−1 are visible. Also the spectra show bands specific to C=O of esters at 1743 and C-O stretching vibration at 1162 cm−1 [24–26].

FTIR spectra of one of the studied oils (corn oil), before and after 36-h heating procedure, a 3850–500 cm−1, b focus on 1850–500 cm−1 range, c focus on 3850–1850 cm−1 range
As it could be seen in Fig. 4, only some small differences are observable on the infrared spectra of the oil sample only in regions around 1050–800, 3450, 2700 and 1650 cm−1 are observable. These differences correspond to the variation in chain length, unsaturation degree and form of the acyl groups [24, 27].
Partial least squares (PLS) and genetic algorithm–PLS regression analysis
Random sample selection was used to form the calibration and validation sets. PLSR model were constructed for estimating the AV, PV and %FAs (C16:0, C18:0, C18:1, C18:2, C18:3), using the 4000–550 cm−1 region of ATR-FTIR spectra. The effect of autoscaling, multiplicative scatter correction (MSC) and orthogonal signal correction (OSC) as the preprocessing methods on the quality of regression were surveyed. The results are shown in Table 1. Also the combination of GA and PLSR were used for wavelength selection. Moreover, the results of autoscaling and MSC as the pretreatment methods of GA–PLS are shown in Table 1.
According to Table 1, the best results were obtained with PLS regression using OSC as pretreatment method for all the dependent parameters (AV, PV, C16:0, C18:0, C18:1, C18:2 and C18:3) in both calibration and validation sets. By comparison of the results of OSC–PLS with other pretreatments combined with PLS (Autoscaling–PLS and MSC–PLS), it can be observed that lower number of LVs was used in case of using OSC. The advantage of model constructed by PLS combined with OSC preprocessing is simplicity (lower numbers of LVs) and good prediction ability. The correlation coefficients of calibration (>0.99) and validation set (>0.86 and in most case >0.94) for all parameters of oils could be considered as excellent statistical performance.
However, OSC–PLS (without wavelength selection) showed the best results in prediction of the desired responses (AV, PV, C16:0, C18:0, C18:1, C18:2 and C18:3). The other developed models (PLS or GA–PLS combined by autoscaling or MSC) represented good ability in most cases. The worst results were belonging to the models used to predict PV. However, as it was mentioned previously, OSC–PLS can be used as a perfect option to predict PV of edible oils based on their FT-IR spectra during heating process.
It is worthy to mention that the amounts (%) of fatty acids (C16:0, C18:0, C18:1, C18:2 and C18:3) were obtained by the GC as the reference method whereas the AV and PV were determined based on titration methods. Therefore, the lower success of most models in prediction of PV can be because of higher error in experimental value of response. The accuracy of iodometric titration of peroxides depends on some other experimental factors such as precise timing and protection of the reaction mixture from oxygen [28]. Also, accurate detection of AV by colorimetric indicator especially for highly colored used oils is difficult [29].
Figure 5 shows the OSC–PLS predicted values of the calibration set for (a) AV, (b) PV, (c) C16:0, (d) C18:0, (e) C18:1, (f) C18:2, (g) C18:3 versus actual amounts and indicates excellent agreement between predicted and actual values.

Predicted values of a acid value, b peroxide value, c C16:0, d C18:0, e C18:1, f C18:2, g C18:3 in the calibration set by OSC–PLS versus their actual amounts
The number of selected variables in GA–PLS depends on the type of responses (AV, PV, C16:0, C18:0, C18:1, C18:2 and C18:3) and was varied between 1213 and 1302. However, the selected wavenumbers were dispersed between the range of 4000–550 cm−1, but there are some wave numbers that were selected for all responses. For example, in MSC GA–PLS model, the wavenumbers of 3353, 2577 and 925 cm−1 were selected for modeling of all seven responses whereas 51 common wavenumbers were selected for modeling of six properties (out of seven studied properties). Because of diversity in the selected wavenumbers, it is hard to make a connection between the modeled responses and type of vibration using the selected wavenumbers.
For assessment of the proposed multivariate method, a comparison was made by other reports. Among the previous reports based on FTIR to determine FA content, the work of Maggio et al. [12] could be noted. They studied only virgin olive oil and by using PLS LV of equal or larger than 13, they obtained RMSEs in the range of 0.002–0.0042. However, our models are simpler (e.g., the number of LVs was 4) and were extended to four types of oils with acceptable RMSEs in range of 0.01–0.08. In addition, the multivariate determination of PV with the use of first derivative of spectrum has been achieved by the Maggio et al. with RMSE of calibration of 0.0143 for normal spectrum and 0.0069–0.0095 for first and second derivative spectrum, respectively. In this case, results of the current work are comparable with their results. Talpur et al. [6] also used single bounce ATR-FTIR for determination of FFA, PV, iodine value, and conjugated diene and triene of one type of oil (cottonseed oil) during potato chips frying for 10 h and obtained RMSE ≤ 0.133 for calibration.
Christy et al. [4] determined C18:1 and C18:2 using second derivative profiles of spectra for eight type of edible oils with excellent Error of Prediction (% w/w) ≤4.1; but their attempt to establish calibration models for C18:3 was not successful. The preference of our present study is obtaining good calibration models for C18:3, C18:0 and C16:0 in addition to C18:1 and C18:2 and simultaneously estimation of the acid value and peroxide value as two important factors for quality control of edible oils.
Conclusion
In this study, we achieved appropriate method for simultaneously determination of %C16:0, C18:0, C18:1, C18:2, C18:3, AV and PV using OSC–PLS as an efficient chemometrics treatment on ATR-FTIR spectra. In this proposed method, seven important factors (noted above) could be easily determined simultaneously by recording only one ATR-FTIR spectra of small amounts of an oil sample (200 µL) with no need of sample preparation and without using organic solvents. The results confirm the capability of OSC–PLS (and in some cases MSC–PLS, autoscaling–PLS and GA–PLS) method to estimate the content of FAs, PV and AV to evaluate oils’ quality. The obtained models in this work allow one to judge the quality of used oils as well. In other words, this method could be introduced as a rapid and economical alternative of the currently common standard methods for identification of the edible oils and checking their quality in food quality control and industry laboratories.
References
Q. Zhang, A.S. Saleh, J. Chen, Q. Shen, Chem. Phys. Lipids 165, 662 (2012)
C.-Y. Ng, X.-F. Leong, N. Masbah, S.K. Adam, Y. Kamisah, K. Jaarin, Vasc. Pharmacol. 62, 38 (2014)
R.P. Gonçalves, P.H. Março, P. Valderrama, Food Chem. 163, 83 (2014)
A.A. Christy, P.K. Egeberg, Chemometr. Intell. Lab. 82, 130 (2006)
C.A. Nunes, Food Res. Int. 60, 255 (2014)
M.Y. Talpur, H. Kara, S. Sherazi, H.F. Ayyildiz, M. Topkafa, F.N. Arslan, S. Naz, F. Durmaz, Talanta 129, 473 (2014)
M.A. Cantarelli, I.G. Funes, E.J. Marchevsky, J.M. Camiña, Talanta 80, 489 (2009)
B. Hemmateenejad, S. Yousefinejad, Anal. Bioanal. Chem. 394, 1965 (2009)
B. Hemmateenejad, S.F. Farzam, N. Mobaraki, J. Iran. Chem. Soc. 11, 1609 (2014)
A. Naseri, B. Ghasemzadeh, K. Asadpour-Zeynali, J. Iran. Chem. Soc. 13, 679 (2016)
M. Shariati-Rad, M. Hasani, J. Iran. Chem. Soc. 11, 471 (2014)
R.M. Maggio, T.S. Kaufman, M. Del Carlo, L. Cerretani, A. Bendini, A. Cichelli, D. Compagnone, Food Chem. 114, 1549 (2009)
http://www.iso.org/iso/iso_catalogue/catalogue_ics/catalogue_detail_ics.htm?csnumber=11558
http://www.iso.org/iso/iso_catalogue/catalogue_ics/catalogue_detail_ics.htm?csnumber=11559
http://www.iso.org/iso/iso_catalogue/catalogue_tc/catalogue_detail.htm?csnumber=44879
S. Wold, M. Sjöström, L. Eriksson, Chemometr Intell Lab. 58, 109 (2001)
G.G. Dumancas, S. Ramasahayam, G. Bello, J. Hughes, R. Kramer, Trend Anal. Chem. 74, 79 (2015)
N. Kumar, A. Bansal, G. Sarma, R.K. Rawal, Talanta 123, 186 (2014)
P. Gemperline, Practical guide to chemometrics (Taylor & Francis, New York, 2006)
S. Yousefinejad, B. Hemmateenejad, Chemom. Intell. Lab. 149, 177 (2015)
D. Broadhurst, R. Goodacre, A. Jones, J.J. Rowland, D.B. Kell, Anal. Chim. Acta 348, 71 (1997)
N. Prieto, P. Oliveri, R. Leardi, M. Gay, C. Apetrei, M. Rodriguez-Méndez, J. de Saja, Sensor Actuat. B Chem. 183, 52 (2013)
Y. Le Dréau, N. Dupuy, J. Artaud, D. Ollivier, J. Kister, Talanta 77, 1748 (2009)
Y. Le Dréau, N. Dupuy, V. Gaydou, J. Joachim, J. Kister, Anal. Chim. Acta 642, 163 (2009)
N. Sinelli, M.S. Cosio, C. Gigliotti, E. Casiraghi, Anal. Chim. Acta 598, 128 (2007)
J. Moros, M. Roth, S. Garrigues, M. de la Guardia, Food Chem. 114, 1529 (2009)
S. Armenta, S. Garrigues, M. De la Guardia, Anal. Chim. Acta 596, 330 (2007)
S. Mahesar, S. Sherazi, A. Khaskheli, A. Kandhroc, S. Uddin, Anal. Methods 6, 4956 (2014)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mahboubifar, M., Yousefinejad, S., Alizadeh, M. et al. Prediction of the acid value, peroxide value and the percentage of some fatty acids in edible oils during long heating time by chemometrics analysis of FTIR-ATR spectra. J IRAN CHEM SOC 13, 2291–2299 (2016). https://doi.org/10.1007/s13738-016-0948-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-016-0948-1