
Abstract
Wiskott–Aldrich syndrome (WAS) is an X-chromosome recessive immunodeficiency disease characterized by the triad of thrombocytopenia, eczema, and susceptibility to infection owing to WAS protein gene abnormalities. Kidney transplantation is rarely offered to WAS patients with end-stage renal disease because of concerns that thrombocytopenia and immune disorders may affect the clinical outcome. Here, we report the case of a 20-year-old kidney transplant patient who developed end-stage renal disease owing to immunoglobulin (Ig)A nephropathy caused by WAS. Despite recurrent IgA nephropathy and T-cell-mediated rejection 7 months after transplantation, two rounds of steroid pulse therapy attenuated his renal function and urinary abnormality. His serum creatinine level was maintained at approximately 1.5 mg/dL 1 year after transplantation. No other WAS-related complications were observed throughout the clinical course. Although WAS can cause poor prognosis in kidney transplant patients, careful follow-up may allow kidney transplantation to be performed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wiskott–Aldrich syndrome (WAS) is an X-chromosome recessive immunodeficiency disease characterized by the triad of thrombocytopenia, eczema, and susceptibility to infection owing to WAS protein (WASP) gene abnormalities [1]. The estimated birth prevalence of WAS is 0.01/1000 births, and over 300 associated gene mutations have been reported [1]. The symptoms and clinical presentation of patients with WASP vary depending on the gene mutations. Some patients develop severe immunodeficiency and die because of hemorrhagic diathesis in their childhood, whereas others have mild thrombocytopenia [1]. It is known that 19% of patients with WAS are complicated by nephropathy, and 70% of these patients have immunoglobulin (Ig)A nephropathy [2]. Although kidney transplantation can be selected as a treatment for end-stage renal disease caused by WAS, there are concerns that immunodeficiency and thrombocytopenia affect renal outcomes [3]. Since the prevalence of WAS is low, and patients who undergo kidney transplantation are extremely rare, the clinical course of kidney transplantation among patients with WAS remains unknown.
Herein, we report the clinical course of a patient with WAS who underwent kidney transplantation as well as literature review.
Case report
A 20-year-old man who wished to receive a kidney transplant was genetically diagnosed with WAS at 8 years of age. He experienced refractory atopic dermatitis and thrombocytopenia at that time. According to the results of the genetic analysis, he and his mother had the same missense mutation associated with WAS (c.713T > C, p.11e238Thr). At 14 years of age, a renal biopsy was conducted to elucidate proteinuria and hematuria etiology, and he was diagnosed with IgA nephropathy (Oxford classification M0E1S0T0C1). Despite aggressive treatments for IgA nephropathy, including several rounds of steroid pulse therapy, tonsillectomy, and administration of cyclosporine and mizoribine, his proteinuria persisted, and his renal function gradually deteriorated and resulted in end-stage renal disease. Besides renal dysfunction, WAS-related symptoms did not require further therapies, such as splenectomy and hematopoietic stem cell transplantation. In addition, no autoantibodies were detected.
At 19 years of age, hemodialysis therapy was initiated because of end-stage renal disease. With careful consideration of the risks of kidney transplantation, an ABO-compatible living-donor kidney transplant was performed with a donated kidney from his father 7 months after the initiation of hemodialysis. The details of the blood examination are shown in Table 1. The total ischemic time was 145 min, and his serum creatinine level gradually declined after transplantation. The baseline transplant kidney biopsies (0 h and 1 h) revealed no significant IgA deposition on the glomeruli and no electron-dense deposits on electron microscopy. Besides severe arteriosclerosis, there were no pathological findings that could affect the graft function. Further, a renal biopsy performed on day 7 postoperatively because of delayed graft function showed no evidence of IgA deposition on the glomeruli or acute rejection. Mesangial proliferation, endothelial hypercellularity, and thrombotic microangiopathy were not observed either; only acute tubular necrosis seemed to be responsible for his reduced renal function. Immunosuppression therapy was conducted as follows: methylprednisolone 4 mg/day, tacrolimus dose adjusted to within 3–5 ng/mL in the trough concentrations, mycophenolate mofetil 1500 mg/day, and everolimus 1.5 mg/day (ceased 7 months after transplantation because of massive proteinuria).
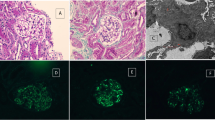
Urinary abnormalities such as hematuria or proteinuria were not observed, and his serum creatinine levels were maintained at approximately 0.9 mg/dL until 4 months after transplantation; however, hematuria and proteinuria began to increase gradually. As his proteinuria increased linearly and reached 2.5 g/gCr, an additional renal biopsy was performed 7 months after transplantation. Nine glomeruli were obtained, three of which showed mild mesangial cell proliferation, and one glomerulus showed fibrinoid necrosis (Fig. 1a). Immunostaining revealed massive but segmental IgA and C3 deposition in the glomerulus (Fig. 1b and Fig. 1c). Despite weak IgM deposition, IgG and C1q depositions were not observed. Electron microscopy indicated electron-dense deposits in the mesangial area (Fig. 1d). In addition to the evidence of IgA nephropathy recurrence, borderline change, tubulitis, and interstitial cellular infiltration were observed (Banff classification t2, i1, g0, v0, ptc0, C4d0) (Fig. 1e). SV-40 staining was negative. Consequently, urinary abnormalities and reduced renal function were attributed to the active IgA nephropathy recurrence and T-cell-mediated rejection, despite the patient not meeting the criteria for T-cell-mediated rejection.

Pathological findings. a Fibrinoid necrosis 7 months after transplantation (Masson Trichrome × 200) (arrow). b Immunoglobulin A deposition on the glomerulus 7 months after transplantation (× 200). c C3 deposition in the glomerulus 7 months after transplantation (× 200). d Electron microscopy 7 months after transplantation. Arrowheads indicate electron-dense deposits in the mesangial area. (× 1200). e Interstitial cellular infiltration and tubulitis 7 months after transplantation (PAS × 100). f Fibrocellular crescent 9 months after transplantation (PAS × 200) (arrowheads)
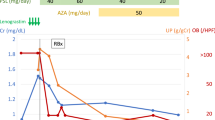
Although the degree of proteinuria declined to 1.2 g/gCr after steroid pulse therapy (methylprednisolone 500 mg × 3), the patient’s serum creatinine level increased to 2.7 mg/dL; additional renal biopsy was performed 9 months after transplantation. Compared with the findings of the previous biopsy, the activity of IgA nephropathy was reduced. However, two glomeruli showed active fibrocellular crescents (Fig. 1f), and the other two glomeruli showed mild mesangial cell proliferation. Although the renal biopsy revealed microvascular inflammation and C4d staining in the peritubular capillary was positive (Banff classification: t1, i1, g1, v0, ptc2, C4d2), there was no definitive evidence of anti-donor antibodies according to the LAB® screen assay. Consequently, we administered steroid pulse therapy to treat the recurrent IgA nephropathy, and the serum creatinine level decreased to 1.5 mg/dL and was maintained at approximately the same level until 1 year after transplantation. Despite slightly decreased IgG and platelet levels, no severe WAS-related symptoms were observed. For example, his IgG level decreased to 705 mg/dL and platelet count decreased to 74,000/μL at 9 months after transplantation. The IgA level was 457 mg/dL before the transplantation, but it decreased to less than 300 mg/dL after transplantation. The clinical course summary of this case is illustrated in Fig. 2, and a summary of the kidney biopsy results is provided in Table 2.

Clinical course. EVR everolimus, Tac tacrolimus, MMF mycophenolate mofetil, mPSL methylprednisolone, Cr serum creatinine, KT kidney transplantation
Discussion
Here, we report about a patient with WAS who underwent kidney transplantation and developed renal dysfunction owing to the early postoperative recurrence of IgA nephropathy. Interstitial lesions were observed on biopsy 7 months after transplantation, suggesting that T-cell-mediated rejection was superimposed on IgA nephropathy in this case. After two rounds of steroid pulse therapy, his renal function and urinary abnormalities resolved.
WAS is an X-chromosome recessive primary immunodeficiency disease with three symptoms of thrombocytopenia, eczema, and susceptibility to infection owing to WAS gene abnormalities. WASP is expressed in the cytoplasms of non-erythroid hematopoietic cells (hematopoietic cells) and plays an important role in remodeling the actin cytoskeleton [4]. Insufficient remodeling of the actin cytoskeleton causes the dysfunction of T cells, B cells, natural killer cells, and antigen-presenting cells. Specifically, the instability in the interaction site between T cells and antigen-presenting cells results in abnormal T cell function. The depletion of B cells is also a known complication of WAS, and the T cell malfunction may disrupt the homeostasis of B cells.
Over 300 WAS gene mutations have been reported to date, and some variants evoke classic WAS that causes severe immune deficiency and death in childhood owing to hemorrhagic diathesis. In contrast, the mild symptom of X-linked thrombocytopenia is observed in some WASP gene mutations [1]. The clinical characteristics of patients with WASP gene mutations vary, and 72% of cases are complicated by autoimmune diseases [5]. Although the association between the gene mutations in this case and the clinical phenotype remains unknown, this patient was diagnosed with WAS owing to repeated upper respiratory tract infections in childhood, thrombocytopenia, and atopic dermatitis. Nonetheless, his phenotype can be assumed to be milder because he grew to be 20 years old.
Since the clinical symptoms vary depending on the cases, general condition should be assessed as an indication for kidney transplantation among patients with WAS. Although the mutation observed in this case (c.713T > C, p.11e238Thr) has not been reported, a past report elucidated that missense mutations tend to be milder phenotypes than nonsense mutations, and mutations in the exon 7 do not cause severe symptoms [6]. Additionally, as shown in Table 1, thrombocytopenia was very mild and IgG level was 1571 mg/dL without any specific treatment for WAS. Consequently, the kidney transplantation was conducted with the consent of the recipient and donor.
Recurrent IgA nephropathy is observed in 7–30% of ordinary kidney transplant patients whose etiology is IgA nephropathy [7]. Generally, recurrent IgA nephropathy tends to be observed in the later post-transplant phase, and the mean timing of recurrence was reported to be 31 months after transplantation [8]; however, some cases develop recurrence soon after transplantation. Some clinical characteristics, such as the development of rapid progressive IgA nephropathy before transplantation [9] and crescents in the intrinsic kidneys [10], are known to be associated with the early recurrence of IgA nephropathy. The current case was diagnosed with IgA nephropathy at 14 years of age, and an active crescent was observed in the biopsy at that time. In addition, he developed end-stage renal disease 5 years after diagnosing IgA nephropathy. Therefore, these characteristics may have played an important role in the early recurrence of IgA nephropathy in this case.
Incidentally, the recurrence of IgA nephropathy in kidney transplant patients with WAS has not been described in detail; however, the pathological features observed in this case may differ from those of other recurrent IgA nephropathy cases. This case exhibited focal segmental necrotizing lesions and crescents in renal biopsies after transplantation, and these findings are not common in kidney transplant patients [11]. Moreover, a previous report indicated that recurrent IgA nephropathy with crescents after kidney transplantation suggested poor renal prognosis because rapid progressive renal failure could be evoked [10]. Even though interstitial lesions can be caused by IgA nephropathy, the interstitial lesions might not have been caused by recurrent IgA nephropathy but rather by chronic rejection in this case. This is because interstitial lesions progressed in a short time. In contrast, IgA nephropathy damages the kidney gradually. Nonetheless, his intrinsic kidney progressed to end-stage renal disease in 5 years; we cannot exclude the influence of IgA nephropathy on the interstitial lesions.
Concerning rejections, T-cell-mediated rejection was previously observed in several kidney transplant patients with WAS [12, 13]. As described above, WAS patients are often complicated with autoimmune diseases, and T cells are involved in the pathophysiology of this disorder [14]. Since regulatory T cells play an important role in T-cell-mediated rejection [15], it should be noted that rejection could occur among transplant patients with WAS. In this case, microvascular inflammation, peritubular capillaritis, and C4d positivity in peritubular capillary were observed in renal biopsy at 9 months after transplantation, suggesting simultaneous existence of antibody-mediated rejection. Although donor-specific antibodies were not detected, anti-non-human leukocyte antigen (HLA) antibody-mediated antibody rejection possibilities could not be excluded. Therefore, plasma exchange or rituximab treatment should have been considered soon after renal biopsy. We attributed the etiology of IF/TA to chronic rejection, probably due to T-cell-mediated rejection. However, non-HLA antibody-mediated rejection might have involved.
Nephropathy caused by WAS can result in end-stage renal disease, and kidney transplantation is a treatment option for this condition. However, kidney transplantation might be avoided in such patients because of the immune deficiency and thrombocytopenia caused by WAS [3]. There are several reports of kidney transplantation in patients with WAS (Table 3). However, the etiologies of end-stage renal disease were not limited to IgA nephropathy among these previous cases.
Furthermore, although immunosuppressive therapy is administered to treat IgA nephropathy in the native kidney, there is no specific established therapy for recurrent IgA nephropathy in transplanted kidneys [16]. In addition, there is no standard treatment for IgA nephropathy caused by WAS. A previous report showed that steroid pulse therapy and tonsillectomy achieved the remission of WAS-induced IgA nephropathy in the intrinsic kidney [17]. Another previous case illustrated the efficacy of steroid therapy in treating recurrent IgA nephropathy in a patient with segmental necrotic lesion and crescents, which were also observed in the current case [18]. In our case, steroid pulse therapy improved the renal outcome. Steroid pulse therapy played a crucial role in treating rejection and the recurrent IgA nephropathy in this case. From the transition of urinary proteinuria severity, glomerular lesions may persist after steroid pulse therapy, suggesting that steroid pulse therapy was more effective on rejection than IgA nephropathy.
However, immunosuppressive therapy should be administered with caution in patients with WAS because immunodeficiency and infection may accelerate abnormal IgA production, which could worsen IgA nephropathy [2]. Furthermore, steroid pulse therapy can induce hypercoagulability, which could cause severe complications, such as venous thrombosis. Fortunately, no severe infections or thrombosis associated with steroid pulse therapy were observed in our case.
The description in this case report had a few limitations. First, everolimus may have affected the degree of proteinuria, although the drug was discontinued to reduce proteinuria. Second, although donor-specific antibodies were not detected at 9 months after transplantation, non-HLA antibodies may have evoked antibody-mediated rejection. Finally, in this case, the condition could be considered mild WAS; however, he developed end-stage renal diseases at the age of 19, so the patient was younger than those in the seven previously reported cases [3, 12, 13, 19,20,21,22]. Therefore, further studies will be needed to elucidate the association between the severity of WAS and age.
In conclusion, we performed ABO-compatible living-donor kidney transplantation for a 20-year-old man. According to accumulating reports, this is the eighth case of kidney transplant in a patient with WAS. Our patient developed recurrent IgA nephropathy and T-cell-mediated rejection. The recurrent IgA nephropathy developed in a shorter period in our case than in previous cases, and our patient was younger than previous patients. Although recurrent IgA nephropathy was pathologically confirmed 7 months after transplantation, the patient’s serum creatinine level was maintained at approximately 1.5 mg/dL 1 year after transplantation. It remains unclear whether kidney transplantation is preferable to conservative treatment in patients with WAS; however, kidney transplantation can be an option for these patients with the proper use of steroids.
References
Malik MA, Masab M. Wiskott-Aldrich syndrome. Treasure Island (FL): StatPearls Publishing LLC; 2021.
Liu C-H, Wu K-H, Lin T-Y, Wei C-C, Lin C-Y, Chen X-X, et al. Wiskott-Aldrich syndrome with IgA nephropathy: a case report and literature review. Int Urol Nephrol. 2013;45:1495–500.
Chovancova Z, Kuman M, Vlkova M, Litzman J. Successful renal transplantation in a patient with a Wiskott-Aldrich syndrome protein (WASP) gene mutation. Transpl Int. 2015;28:1005–9.
Blundell MP, Worth A, Bouma G, Thrasher AJ. The Wiskott-Aldrich syndrome: the actin cytoskeleton and immune cell function. Dis Markers. 2010;29:157–75.
Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, et al. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. 2003;111:e622–7.
Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, et al. Mutations of the Wiskott-Aldrich syndrome protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood. 2004;104:4010–9.
Blosser CD, Bloom RD. Recurrent glomerular disease after kidney transplantation. Curr Opin Nephrol Hypertens. 2017;26:501–8.
Bumgardner GL, Amend WC, Ascher NL, Vincenti FG. Single-center long-term results of renal transplantation for IgA nephropathy. Transplantation. 1998;65:1053–60.
Otsuka Y, Takeda A, Horike K, Inaguma D, Goto N, Watarai Y, et al. Early recurrence of active IgA nephropathy after kidney transplantation. Nephrology. 2014;19:45–8.
Park S, Baek CH, Cho H, Yu MY, Kim YC, Go H, et al. Glomerular crescents are associated with worse graft outcome in allograft IgA nephropathy. Am J Transplantat. 2019;19:145–55.
Floege J, Grone HJ. Recurrent IgA nephropathy in the renal allograft: not a benign condition. Nephrol Dial Transplant. 2013;28:1070–3.
Meisels IS, Strom TB, Roy-Chaudhury P, Abrams J, Shapiro ME. Renal allograft rejection in a patient with the Wiskott-Aldrich syndrome. Transplantation. 1995;59:1214–5.
Fischer A, Binet I, Oertli D, Bock A, Thiel G. Fatal outcome of renal transplantation in a patient with the Wiskott-Aldrich syndrome. Nephrol Dial Transplant. 1996;11:2077–9.
Maillard MH, Cotta-De-Almeida V, Takeshima F, Nguyen DD, Michetti P, Nagler C, et al. The Wiskott-Aldrich syndrome protein is required for the function of CD4+CD25+Foxp3+ regulatory T cells. J Exp Med. 2007;204:381–91.
Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–52.
Lionaki S, Panagiotellis K, Melexopoulou C, Boletis JN. The clinical course of IgA nephropathy after kidney transplantation and its management. Transplant Rev (Orlando). 2017;31:106–14.
Kakio Y, Uchida HA, Kitagawa M, Arata Y, Kato A, Inoue-Torii A, et al. IgA nephropathy complicated with X-linked thrombocytopenia. Acta Med Okayama. 2018;72:301–7.
Tomiyoshi Y, Sakemi T, Ikeda Y, Ohtsuka Y, Nakamura M, Fujisaki T. Cellular crescents and segmental glomerular necrosis in IgA nephropathy are indicative of the beneficial effects of corticosteroid therapy. Intern Med. 2001;40:862–6.
Webb MC, Andrews PA, Koffman CG, Cameron JS. Renal transplantation in Wiskott-Aldrich syndrome. Transplantation. 1993;56:1585.
Garnier AS, Augusto JF, Pellier I, Subra JF, Sayegh J. Successful long-term outcome of kidney transplantation in a patient with X-linked thrombocytopenia: 9 year follow-up. Transplantation. 2014;98:e57–8.
Kai K, Sumida M, Motoyoshi Y, Ogawa Y, Miki K, Iwadoh K, et al. Successful long-term graft survival of a renal transplantation patient with Wiskott-Aldrich syndrome. Intern Med. 2016;55:1761–3.
Al Midani A, Donohue C, Berry P, Jones G, Fernando B. Use of thromboelastography to guide platelet infusion in a patient with Wiskott-Aldrich syndrome undergoing renal transplant. Exp Clin Transplant. 2020;18:636–7.
Acknowledgements
The pathological findings of this case were diagnosed by Dr. Takashi Taguchi and Dr. Satoshi Hisano. Unfortunately, both have since passed away.
Funding
The authors received no specific funding for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
The present report and all procedures described in it have been carried out in accordance with the 1964 Declaration of Helsinki and its later amendments.
Informed consent
The patient provided a signed informed consent form for publication of the case report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Yamaguchi, K., Kitamura, M., Kawaguchi, Y. et al. A case of early recurrent immunoglobulin A nephropathy and T-cell-mediated rejection in a transplant patient with Wiskott–Aldrich syndrome. CEN Case Rep 11, 60–66 (2022). https://doi.org/10.1007/s13730-021-00631-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-021-00631-9