
Abstract
Anticoagulant-related nephropathy (ARN), a significant but frequently undiagnosed problem in patients receiving anticoagulation, is found to be associated with increased renal morbidity and all-cause mortality. While ARN is mainly associated with warfarin use, recent case reports suggest that it may also occur in patients taking direct oral anticoagulants (DOAC). We report a patient who had a history of alcoholic liver cirrhosis and paroxysmal atrial fibrillation, and received dabigatran 110 mg twice daily for 1 year. He presented with gross hematuria and severe acute kidney injury with an international normalized ratio of 4.09. Dabigatran was stopped and he was put on temporary hemodialysis support. His renal function gradually improved when the hematuria subsided. Renal biopsy later confirmed the presence of red blood cell casts inside the renal tubules with features of IgA nephropathy. Finally, his renal function returned back to baseline level. As DOAC has been increasingly used nowadays for the treatment of various thromboembolic diatheses, regular monitoring of renal function is warranted, especially in patients with underlying glomerular diseases and coagulopathy such as chronic liver diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anticoagulant-related nephropathy (ARN) is a significant but frequently undiagnosed problem in patients receiving anticoagulation. It is found to be associated with increased renal morbidity and all-cause mortality. However, the paucity of renal biopsy data made it difficult to estimate the exact incidence and renal outcomes of ARN. A presumptive definition of ARN is acute kidney injury (AKI) without other obvious etiologies in the setting of an international normalized ratio (INR) of > 3 [1]. However, the definitive diagnosis of ARN should be made by renal biopsy. Renal tubular lumen obstruction with red blood cell (RBC) casts leading to acute tubular necrosis (ATN) is rendered the key histological hallmark [2]. While ARN is mainly associated with warfarin use, direct oral anticoagulants (DOAC) may be risk factors as well, especially in patients with underlying chronic kidney disease, hypertension and diabetes mellitus [1]. This topic is important nowadays, because DOAC are increasingly being used to treat thromboembolism and for stroke prophylaxis in patients with atrial fibrillation. Here we report a patient with underlying alcoholic liver cirrhosis and paroxysmal atrial fibrillation receiving dabigatran for 1 year, presented with markedly elevated INR, macroscopic hematuria and severe AKI. His renal function significantly improved when the coagulopathy was corrected by vitamin K.
Case history
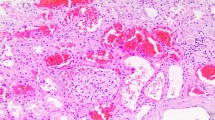
A 61-year-old man with alcoholic liver cirrhosis (Child–Pugh Score B), hypertension, paroxysmal atrial fibrillation on dabigatran 110 mg twice daily for 1 year, presented with gross hematuria with AKI. He did not have baseline proteinuria or microscopic hematuria. He did not receive any angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, nonsteroidal anti-inflammatory drugs or herbal medicine. His appetite was good. On admission, the blood pressure was 144/86 mmHg. Laboratory results included serum creatinine (SCr) 418 umol/L (baseline SCr 87 umol/L), prothrombin time (PT) 45.4 s with an INR of 4.09, activated partial thromboplastin time (APTT) 68.3 s, hemoglobin 11.7 g/dL, platelet count 73 × 109/L and 24-h urine protein 3.04 g. Autoimmune markers including anti-neutrophil cytoplasmic antibodies and anti-glomerular basement membrane antibody were negative. The serum complement level and immunoglobulin level were normal. Urine microscopy did not show any RBC casts. Renal ultrasound, computerized tomography urogram and cystoscopy did not reveal any abnormality. Dabigatran was stopped immediately and intravenous vitamin K was started at a dose of 10 mg once daily. However, his SCr kept creeping up to more than 900 umol/L. In view of the presence of uremic symptoms, temporary hemodialysis support was started 1 week after hospitalization. Renal biopsy was postponed initially due to the presence of coagulopathy. His clotting profile improved after given vitamin K (PT 16 s, INR 1.36 and APTT 39.3 s after 2 weeks). The gross hematuria gradually subsided, followed by progressive improvement in renal function. He did not require further dialysis treatment after 3 weeks. Renal biopsy was subsequently performed after the complete resolution of gross hematuria (at 4 weeks after admission). Light microscopy revealed moderate interstitial inflammation and diffuse ATN with RBC casts inside the renal tubules (Fig. 1). Of the 27 glomeruli, one showed cellular crescent formation. Fibrinoid necrosis and fibrous crescent were not found. Indirect immunofluorescence showed diffuse global 3+ granular mesangial and capillary loop staining with IgA and C3. There was linear accentuation of IgG along the glomerular basement membrane (GBM) with focal segmental non-specific trapping of C1q, while there was no significant glomerular deposit of IgM. Electron microscopy revealed the presence of some subendothelial electron dense deposits. There was no abnormal GBM thinning or multilamination. These features support a diagnosis of IgA nephropathy (IgAN) with MEST-C score E1, M0, S1, T0, C1. At that time, his SCr further improved to about 300 umol/L. In view of the histological presence of acute tubulointerstitial inflammation, prednisolone (0.5 mg/kg/day) was started 5 weeks after hospitalization and his SCr finally returned back to baseline level 4 months after the acute event.

Light microscopy (trichrome stain) showing moderate interstitial inflammation and diffuse acute tubular necrosis with red blood cell casts inside the renal tubules
Discussion
Glomerular hematuria, a well-established manifestation of glomerular diseases of various etiologies, has been shown to increase the risk of AKI and adversely affect the long-term renal outcome [3]. IgAN, thin-membrane disease, and Alport syndrome are frequent causes of macroscopic glomerular hematuria where hematuria by itself was traditionally considered an innocuous consequence of disease manifestation rather than a culprit leading to kidney damage. Praga et al. reported that renal impairment was observed in 38% episodes of macroscopic hematuria in patients with IgAN [4], a risk which increased after the use of anticoagulant. In 25% of patients with IgAN and AKI, their renal function did not fully recover and macroscopic hematuria > 15 days was the only statistically significant prognostic factor [5]. Postulated mechanism of ATN during episodes of macroscopic hematuria include mechanical obstruction by RBC casts, oxidative stress imparted by hemoglobin, heme or free iron, and depletion of nitric oxide leading to intrarenal vasoconstriction and ischemia [3].
Coagulopathy, either therapeutically induced or intrinsic due to disease processes such as liver failure, exacerbates the tendency of glomerular hemorrhage in patients with pre-existing glomerular diseases. Glomerular hemorrhage and ATN have been observed in patients with warfarin overdose, while active glomerulonephritis was excluded by renal biopsy [6]. An INR > 3 both increases the risk of AKI and accelerates the progression of chronic kidney disease [1]. Even a moderate increase in INR has been shown to aggravate macroscopic hematuria in patients with IgAN leading to AKI [7]. Recently, it has been shown that ARN is not only observed in patients with warfarin but also in those receiving DOAC [8,9,10,11,12]. In our patient, the coagulopathy could be potentiated by the underlying liver cirrhosis in addition to dabigatran.
The mainstay of treatment of ARN is largely supportive, which includes the correction of coagulopathy. It is possible that prolonged coagulopathy can lead to continued glomerular hemorrhage and additional kidney tubular damage [13]. There are case reports demonstrating that the addition of idarucizumab for patients with AKI due to dabigatran can result in more rapid improvement of renal function [12]. On the other hand, the use of steroid to treat AKI due to ARN is largely anecdotal. Patients with more prominent crescent formation and tubulointerstitial inflammation should be considered to be candidates for steroid therapy, aiming at attenuating the inflammatory response and facilitating renal recovery. As a result, despite the fact that the renal function in our patient already improved significantly with the resolution of hematuria, steroid therapy was also indicated for the treatment of underlying IgAN. Concerning the prognosis, our patient, like most of the cases of dabigatran-induced AKI, could achieve full recovery of renal function [12].
In conclusion, DOAC has been increasingly used nowadays for the treatment of various thromboembolic diatheses. Since the data on the safety of DOAC with regard to renal toxicity and incidence of ARN are still limited, regular monitoring of renal function for early detection of ARN is warranted, especially in patients with underlying glomerular diseases and coagulopathy such as chronic liver diseases.
References
Brodsky SV, Nadasdy T, Rovin BH, et al. Warfarin-related nephropathy occurs in patients with and without chronic kidney disease and is associated with an increased mortality rate. Kidney Int. 2011;80:181–9.
Delclaux C, Jacquot C, Callard P, Kleinknecht D. Acute reversible renal failure with macroscopic haematuria in IgA nephropathy. Nephrol Dial Transpl. 1993;8:195–9.
Moreno JA, Martín-Cleary C, Gutiérrez E, et al. AKI associated with macroscopic glomerular hematuria: clinical and pathophysiologic consequences. Clin J Am Soc Nephrol. 2012;7:175–84.
Praga M, Gutierrez-Millet V, Navas JJ, et al. Acute worsening of renal function during episodes of macroscopic hematuria in IgA nephropathy. Kidney Int. 1985;28:69–74.
Gutiérrez E, González E, Hernández E, et al. Factors that determine an incomplete recovery of renal function in macrohematuria-induced acute renal failure of IgA nephropathy. Clin J Am Soc Nephrol. 2007;2:51–7.
Brodsky SV, Satoskar A, Chen J, et al. Acute kidney injury during warfarin therapy associated with obstructive tubular red blood cell casts: a report of nine cases. Am J Kidney Dis. 2009;54:1121–6.
August C, Atzeni A, Koster L, Heidenreich S, Lang D. Acute renal failure in IgA nephropathy: aggravation by gross hematuria due to anticoagulant treatment. J Nephrol. 2002;15:709–12.
Moeckel GW, Luciano RL, Brewster UC. Warfarin-related nephropathy in a patient with mild IgA nephropathy on dabigatran and aspirin. Clin Kidney J. 2013;6:507–9.
Escoli R, Santos P, Andrade S, Carvalho F. Dabigatran-related nephropathy in a patient with undiagnosed IgA nephropathy. Case Rep Nephrol 2015;2015:298261.
Gois M, Azevedo A, Carvalho F, Nolasco F. Anticoagulant-related nephropathy in a patient with IgA nephropathy. BMJ Case Rep. 2017. https://doi.org/10.1136/bcr-2016-218748.
Kalaitzidis RG, Duni A, Liapis G, et al. Anticoagulant-related nephropathy: a case report and review of the literature of an increasingly recognized entity. Int Urol Nephrol. 2017;49:1401–7.
Awesat J, Sagy I, Haviv YS, et al. Dabigatran-induced nephropathy and its successful treatment with Idarucizumab—case report and literature review. Thromb Res. 2018;169:120–2.
Wheeler DS, Giugliano RP, Rangaswami J. Anticoagulation-related nephropathy. J Thromb Haemost. 2016;14:461–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare no competing interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, formal consent is not required.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Li, X., Cheung, C.Y. Dabigatran causing severe acute kidney injury in a patient with liver cirrhosis. CEN Case Rep 8, 125–127 (2019). https://doi.org/10.1007/s13730-019-00378-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-019-00378-4