
Abstract
The process to identify genes responsible for a disease is a complex task. The various experimental techniques developed to identify disease-causing genes suffer from the problem of high-cost and high time consumption. Thus, with the increasing amount of biological information available online various computational techniques have been developed to complete this complex task of identification of disease-causing genes. A more accepted view is that the genes related to similar diseases reside in the same neighborhood of the molecular network. In this review, various categories of computational techniques for disease gene prioritization have been highlighted and compared. The work majorly focuses on various categories of approaches that use protein–protein interaction networks with data from heterogeneous sources and heterogeneous biological types. Furthermore, a comparison of these approaches is done and also some issues related to them are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The advancement in technology has uncovered various truths about life. Bioinformatics is one such field that uses advances in the areas of information technology, computer science and communication technology to handle the complicated problems of life sciences, more specifically in biotechnology. Thus, using computational techniques to solve such problems has fast-forwarded the process.
Human beings are made up of genes that are responsible for the functionality and characteristics of the individual. Human being acquires a total arrangement of qualities from each parent, and additionally a tremendous exhibit of social and financial encounters from his/her family. Since relatives speak to most firmly comparable genomic associations and share the same natural encounters along these lines’ family history is frequently observed as one of the most grounded elements for some diseases, for example, cardiovascular diseases, diabetes, cancer, and so on.
Uncovering these disease-causing genes among the candidate genes is thus a major step towards the pathogenesis of diseases. In research after some point in their analysis researchers filter which genes to consider for further experimentation and which one to drop. Disease gene prioritization aims at selecting these disease-causing genes among the large pool of candidate genes by computational examination of both open and private genomic information. Its principle objective is to maximize the yield by narrowing down the gene set to be considered and thus helping in focus on the most promising candidate genes. Thus, disease gene prioritization helps determine the functionality of genes, early diagnosis of disorders and designing effective strategies for treatment.
Disease gene prioritization provides various benefits such as:
-
It can help in the development of tools that will provide personalized information to the individual about the risk of developing a certain disease.
-
It helps health professionals to design prevention programs for each person according to their mark-up.
-
It allows effective selection of the treatments for the patients which are most likely to help them without causing any adverse reactions.
Identifying disease-causing genes and the functionality of various components of genes is a key challenge. Various high-throughput techniques such as genome wide association studies (GWAS) (Hirschhorn and Gadjos 2011), (Ott et al. 2011), linkage analysis, large-scale RNA interference screens (Boutros and Ahringer 2008) are being developed to distinguish disease-causing genes but these mostly resulted in a long list of candidate genes. Subsequently, these experimental methodologies are very time-consuming and expensive as they require several resources. For these various computational systems are being produced for prioritization of hopeful genes. These strategies work by performing estimations iteratively and some additionally coordinate complex, heterogeneous information sets, for example, sequence information, expression information, functional annotation, and other biomedical writing to give a rundown of organized gene list which would be useful for future review in a more educated manner.
In this review, different computational techniques have been discussed with the main attention on network-based disease gene prioritization. In the second section, various types of disease gene prioritization have been discussed. In the third section, various categories of network-based techniques are highlighted; with integration with heterogeneous data types discussed in the fourth section. In the fifth section of the paper, the workflow of the network-based disease gene prioritization process has been explained. In the sixth section discussion about techniques and problems that arise in the gene prioritization process are discussed.
2 Disease gene prioritization techniques
On the basis of strategy and type of data used, various computational techniques for candidate gene prioritization can be extensively classified into the under-mentioned categories: filtering-based techniques, similarity-based techniques, and network-based techniques. Table 1 lists some of the advantages and disadvantages of these techniques.
2.1 Filtering-based techniques
Filtering-based techniques work on the basis of the properties of genes. The properties of genes are defined and then filters are created. With the application of each filter, the list of candidate genes gets reduced leaving at the end the smaller output list of candidate genes that have a higher probability of being linked with the disease. For example, a tool named ‘TEAM’ (Tool for the integration of Expression, and linkage and Association Maps) (Franke et al. 2004) have three-level filtering options for analysis of genes: candidate gene region analysis, whole chromosome analysis, and gene-specific analysis. Filtering-based techniques are also used in combination to other techniques for narrowing down the list of most promising candidate genes. GeneDistiller (Seelow et al. 2008) is a web-based application which offers a customizable prioritization of genes as per user requirements. The user can read the different kinds of information about the genes, select specific genes by applying some filters, sort the genes on basis of parameters and also prioritize genes as per required specification. For the prioritization task, the user can also assign different weights to different parameters considered while calculating the final score. But sometimes due to the strict filtering mechanism, filtering-based techniques sometimes also filter out the genes only if one criterion is not met thus producing false negatives.
2.2 Similarity-based techniques
Similarity-based techniques perform prioritization by calculating the similarity between candidate genes and seed genes. The similarity is identified either on the basis of disease-related keywords searched from disease-related documents or on the basis of similarity between properties of genes. Yu et al. (2008) have investigated various vocabularies and representations and used various ranking algorithms to perform prioritization. Perez-Iratxeta et al. (2002) developed a scoring system using data mining to prioritize genes using information from Medical Subject Headings (MeSH) (Lowe and Barnett 1994). The authors used information from MEDLINE and NCBI RefSeq database to derive relationships among functional terms, chemical terms and pathological conditions. A software named ENDEAVOUR (Aerts et al. 2006) performs gene prioritization on the basis of similarity with the training properties obtained from various data sources individually and then combining the results of all the ranks obtained from different sources into one list. Adie et al. (2006), developed a web-based server SUSPECT which uses the weighted combination of sequence-based and annotation-based approaches to prioritize genes. van Driel et al. (2006) used text mining to calculate disease similarity information by calculating the cosine of angle between the vector representation of each Online Mendelian Inheritance In Man (OMIM) record. In Schlicker et al. (2010) the authors introduced an approach MedSim, which uses functional comparisons to rank genes. The approach particularly automatically annotates GO terms with disease entries to create functional profiles of genes.
Over the time, various methods to calculate similarity between genes and diseases have been developed. Some these methods measure similarity on the basis the probability of the appearance of GO terms, their distances to the closest common ancestor term, the kappa statistics, cosine similarity, and many others. Pesquita et al. (2009), the authors have reviewed various methods used in biomedicine to calculate the similarity and provided the categorization, comparative assessment and various implications of the methods. Although the text mining-based approaches have performed well but they experience the ill effects of the issue of absence of steady representation or association of key ideas which often lead to redundant data. In addition, for the well characterized diseases, i.e., those diseases for which association of genes have already been identified in the past, there is more information retrieval about them using text mining techniques as compared to more newly identified diseases.
2.3 Network-based techniques
A network-based technique primarily uses the topology of the network for ranking the genes. In these techniques, the interactions among the proteins or genes in the organisms are represented using graphs, where hubs represent the entities and the edges represent the connections among them. This graphical representation helps in the visualization of the network which enhances the understanding of the complete biological structure. Various pieces of evidence propose that practically related qualities prompt to comparative phenotype (Oti and Brunner 2007). In addition, many studies have suggested that the genes responsible for the same or comparative diseases are generally located in one of the same biological modules such as in a pathway (Wood et al. 2007), protein complex (Lage et al. 2007), or a subnetwork of protein interactions (Lim et al. 2006). Thus, using these biological modules to understand the cause of the disease is quite helpful. The set-back to such techniques is the scarcity of knowledge of the disease which limits its progress.
3 Network-based disease gene prioritization
Based on their structure and properties, biological networks are divided into three types (Morris et al. 2015a): pathways, interaction networks, and similarity networks. While pathways are used to represent metabolic, signaling or regulatory pathways; interaction networks illustrate the interactions among biological products like genes, amino-acid residues, proteins, metabolites, small molecules, diseases, etc. for example gene regulation networks, metabolic networks, protein interaction networks, and signal transduction networks. In similarity type, nodes represent some biological entities, while edges represent the relation among nodes on the basis of some similarity measure forming networks like metabolic correlation networks using profiling data (Weckwerth et al. 2004), gene or transcript correlation network using expression data (Bergmann et al. 2004) or etc. Amongst the above discussed biological modules, it has been noticed that Protein–Protein Interaction Networks (PPINs) can possibly distinguish the disease-causing genes as these add to a better understanding of disease pathways (Safari-Alighiarloo et al. 2014).
A Potein–Protein Interaction Network (PPIN) is often abstracted as a graph model, where G (V, E) is an undirected graph with a set of nodes-V denoting proteins and set of edges-E denoting interactions among the proteins. Thus determining the properties of genes using the interactions, is what became the basic building component for the development of computational approaches for disease gene prioritization. These approaches work on the principle of “guilt-by-association” (Oti and Brunner 2007) according to which the genes that are functionally or physically related to disorders are more likely to be positioned closer to each other. Thus, on the basis of proximity to disease genes the likelihood of candidate genes being related to the disease can be computed.
Protein–protein interaction networks model physical interactions and functional relationships among the proteins. With the principle objective to identify protein interactions various large-scale experiments have also been conducted such as a high-throughput yeast two-hybrid (Y2H) system (Ito et al. 2000) which discover direct binary interactions among the genes, tandem affinity purification followed by mass spectrometry (Morris et al. 2014). In addition, various text mining applications that provide manually validated and/or computationally predicted PPIs from biological data are available for public use (Blaschke et al. 1999). Thus large-scale identification experiments lead to the generation of billions of interactions and these interactions have been managed in databases such as DIP (Xenarios et al. 2002), BIND (Bader et al. 2003), STRING (Szklarczyk et al. 2011), MINT (Licata et al. 2012), etc. This interaction information is further used to construct a PPI network. Since visualizing the interactions and network is a difficult task various bioinformatics tools such as GenMAPP (Dahlquist et al. 2002), Osprey (Breitkreutz et al. 2003), VisANT (Hu et al. 2005), CellDesigner (Funahashi et al. 2003), PIANA (Aragues et al. 2006), Cytoscape (Cline et al. 2007), Shannon et al. (2003) have been developed along with the various plug-ins. These tools have made the work of the researchers easier. Despite the incompleteness and biased nature of PPI data, PPI networks recently due to its high-coverage and high-quality are one of the most widely used biological networks in predicting and detecting protein complexes (Stelzl et al. 2005). Various topological parameters can be used to identify the collective, subnetwork and individual behavior of the network components. On the basis of topological parameters, the network-based prioritization techniques are broadly classified into local measures-based techniques and global measures-based techniques.
3.1 Local measures-based techniques
The early methods in gene prioritization focused on local network information of the network. Table 2 contains a list of some of the local measures which can be used for analyzing networks.
The most straightforward approach is based on the interconnectedness of the genes, i.e., whether the given genes are connected with disease genes directly in the PPI network. This can be determined either using a direct-neighborhood approach or shortest path approach. Krauthammer et al. (2004) proposed molecular triangulation as a method to identify genes responsible for genetic variations. The authors used text-mining approaches to extract interactions from research literature available online. Each node was initially assigned a primary-evidence score which was then projected to nearest neighbors. The evidence score of a node is the summation of near neighbors’ secondary-evidence score received from nearest neighbors. This evidence score was further used to identify the genes, as the gene which is nearer to more seed genes will have a higher score. Oti et al. (2006) used the direct-neighborhood based approach for predicting disease genes by counting the number of first-degree candidate genes known to be linked with disease genes.
But direct-neighborhood based approaches failed to take into account the fact that the disease genes are also the part of same pathways. Dezső et al. (2009) proposed an algorithm that builds a shortest-path network which connects condition-specific disease genes. The authors calculated the significance score of each node on the basis of the number of paths from the particular node in the shortest path network to the total paths in the complete network. Xi et al. (2016) compared the performance of three different human PPI networks from three different interaction sources that are literature-curated (LC), experimentally derived (EXP) and predicted PPIs. The authors used five topological features namely node degree, 1 N index, 2 N index, the average distance to known disease gene and positive topological coefficient to train a k-Nearest Neighbor (kNN) classifier to predict the genes with a higher likelihood of being involved with the disease. Hsu et al. (2011) considered two factors: one direct interaction between two genes and the second number of connectors between genes for calculating the interconnectedness score of the gene on the basis of which genes were prioritized. Zhu et al. (2012) formulated a vertex-similarity-based approach based on the similarity between two genes. The authors had used both direct neighborhood and shortest path approach to calculate the similarity score between two genes. Li et al. (2014) proposed a simple localized algorithm SPranker which performed prioritization on the basis of topological similarity calculated on the basis of the shortest path from the disease gene. Table 3 contains the list of summary of local measures-based techniques.
3.2 Global measures-based approaches
While the local measures consider only the neighborhood of the node, the global measures consider the complete topology of a network for ranking genes. Various techniques based on global measures have been proposed by researchers to perform disease gene prioritization some of which are discussed as follows.
3.2.1 Graph centrality
Wang et al. (2011) proposed to integrate different overlapping disease signatures on the basis of graph centrality. The authors used six graph centrality measures listed in Table 4 to quantify the importance of a gene in disease, which is along with their definitions on the basis of which prioritization was performed. Higher the value of the centrality measure more is the probability of a gene being related to the disease.
3.2.2 Random walk with restart (RWR)
A random walk on graph is defined as a transition from the present node to a randomly chosen neighbor iteratively starting at a given source node, s. Random walk with restart is a variant of random walk in which the walk is restarted at node every s time step with probability r following equation:
where \(W\) represents a column-normalized adjacency matrix and \(p^{\left( t \right)}\) is a vector, where the ith element of the vector represents the probability of being at node i at time step t and \(p^{\left( 0 \right)}\) is the initial probability vector.
Köhler et al. (2008) defined the similarity between the genes in the network using RWR. The gene ranking was done on the basis of the steady-state probability vector obtained after the change between the values of two iterations is very small (below 10−6). Erten et al. (2011) proposed a gene prioritization scheme on the basis of topological similarity in which the global signature of the location of gene is used to perform disease gene prioritization. The topological profile was constructed using the proximity of protein to every other protein in the network. The authors assessed topological similarity using random walk proximity as a feature. They proposed three different schemes on the basis of topological similarity: average topological similarity with seed genes (ATS); topological similarity with average profile of seed genes (TSA); topological similarity with the representative profile of seed genes (TSR).
Li and Li (2012) proposed two different algorithms based RWR using multigraphs. A multigraph is a graph constructed using different gene networks from multiple data sources and then merging them forming a one single network. The first algorithm RWRM (random walk with restart for multigraphs) simply extends random walk with restart on multigraphs. The second algorithm complex heterogeneous network works by combing multigraph gene network and phenotype information and then performing random walk. Le and Kwon (2013) tried to further enhance the performance of RWR based methods by weight adjustment technique in which weight reinforcement of interactions close to known disease genes is done.
3.2.3 Diffusion kernel
The diffusion kernel K of a graph is defined as
where \(\beta\) is the magnitude of diffusion and L is Laplacian of the graph.
Köhler et al. (2008) used the method of diffusion kernel to rank genes on the basis of the score computed using the following formula:
Zhang et al. (2011) proposed a Bayesian regression approach that uses diffusion kernels on PPI networks that prioritize genes on the basis of Bayes factor which calculates the strength of similarity between disease phenotypes using the gene proximity profile containing pairwise similarity between every pair of genes.
3.2.4 Network propagation
Vanunu et al. (2010) proposed a method, PRINCE which uses prioritization function following logistic regression model-based approach to assigning confidence scores to the genes. The method is similar to RWR with the major difference of using weight matrix which is derived from the adjacency matrix. The key idea was to normalize each edge weight by the degrees of both of its endpoints in contrast to the random walk with restart which considers only one endpoint.
3.2.5 Page rank with priors
Page Rank with Priors (PRP), another algorithm that is used for gene prioritization is an extension of Page Rank. In this algorithm, a prior probability is assigned to each node and further with some probability factor it chooses amongst its neighbors which node to move next. The iterative equation (Gonçalves et al. 2012) it uses is as follows:
where \(\beta\) is back probability denoting the probability of jumping to initial node at each step. Chen et al. (2009) used page rank with priors to rank genes based on the set of known disease genes. Sinsha and Chitturi (2016) highlighted the modified versions of the page rank algorithm to deal with the existing problems of gene prioritization such as bias nature towards neighbors of high degree nodes and central nodes.
3.2.6 Topology potential
Li et al. (2015) proposed a gene prioritization method by considering each protein as a material particle. Each particle around itself generates a potential field and all the interaction and thus results in the formation of a topological field over the entire network. The authors ranked the proteins on the basis of their topology potential value and selected the top x % proteins to identify essential genes as per the requirement. The authors also integrated other centrality measures with topology potential to enhance the performance of the system.
3.2.7 Machine learning
Machine learning has also been used in the field of disease gene prioritization. Lui et al. (2016) defined a Generalized Bi-relational Network (GBN) and used a series of common structure-based features named number of neighbor (NG), Jaccard coefficient, common neighbors (CN), neighbor of neighbors (NN), total neighbors (TN), Adar similarity(Adar), friends measure(FM), cosine similarity, preferential attachment score(PAS), Katz measure for classes of path weighting features. Common features, 4 classes of path weighting features and union of all features were used to compare of supervised-learning models: logistic regression, decision tree (DT), random forest (RF), and K-Nearest neighborhood (KNN). All path weighting features were also calculated using the unsupervised model. Lui et al. (2020) have applied the popular technique, i.e., machine learning for network-based classification. The authors have used supervised learning using the neighborhood of each gene’s network as its feature vector using the influence matrix, adjacency matrix, and node-embedding matrix.
3.2.8 Soft-computing techniques
Grewal et al. (2016) proposed to assign different weightage to different measures and thus used aggregation operators. Aggregation combines different values into a single value which has a combined effect of all the values. The authors extracted values of four different measures, assigned each a fuzzy association score and then combined them using aggregation operators. The aggregation score finally computed for each gene was used to form a prioritization list.
3.2.9 Community detection
Indulekha et al. (2018) presented an algorithm based on community detection in a PPI network to perform the task of gene prioritization. The main idea followed is the identification of community in a graph which is a collection of closely connected nodes. These detected communities are used in analyzing the properties of the network. The authors have used Gervan–Newman (2004) algorithm for the process of community detection which works using the edge betweenness parameter.
Table 5 contains a summary of global measures-based techniques.
4 Integration of heterogeneous data types for disease gene prioritization
The performance of a prioritization technique is heavily influenced by the data provided to the system, because if the seed gene set is too small, the detected pattern is insufficiently informative. With the increasing availability of data, the focus has shifted from using single omics data to integrating it with more heterogeneous data. This multi-omics approach allows better and deeper understanding of the complexity of phenotypes and also helps deal with biasness and missing nature of data. Using different types of biological information related to a disease enhances the knowledge about the disease and has reported to improve the performance of prioritization techniques. Thus, researchers proposed combining protein network information with different types of biological data.
There are mainly two approaches which are followed for the process of integration of data. The first approach involves consideration of data from different sources separately while accessing the genes and then finally aggregating the resultant multiple ranked lists into one final ranking list. The integration of lists is done using one of the many integration techniques like N-dimensional Order Statistic (NDOS) (Stuart et al. 2003), discounted rating system (DRS) (Li and Patra 2010), kernel based fusion (Yu et al. 2011), etc. The alternate approach is integrating the information from various data sources into a single network. A comprehensive network with all types of data is constructed in which different layer contains different type of data and are connected to each other through gene-phenotypic associations, and then the task of prioritization is performed. Various publicly accessible databases like OMIM (Amberger et al. 2019) provides phenotypic–genotypic relationships in human. In Zhang et al. (2019), the authors have constructed quadruple layer heterogeneous network to perform gene prioritization.
4.1 Gene ontology
The gene ontology gives controlled vocabularies of characterized terms speaking to quality gene properties. These cover three spaces: cellular component, molecular function, and biological process. It is organized as a coordinated non-cyclic chart, where every term has characterized connections to at least one different term in a similar space, and now and then to different areas. Li et al. (2014) also proposed another algorithm, i.e., SPGOranker, an improvement over SPranker which uses GO term similarity. The improved algorithm integrates the molecular functional similarity with the SPranker which works on the shortest distance method. The validation results obtained show the improvement in detecting true disease-causing genes with the integration of GO annotations.
Gentili et al. (2019) have proposed two heuristics using similarity-based on Gene Ontology-Biological Node Relevance (BNR) and Biological Random Walk (BRW). In the former technique, the authors have sorted genes on the basis of the score calculated as an intersection between biological information and annotations. BRW is a majorly RWR with the main difference that in BRW the matrices calculated depend on the biological information of the available genes.
4.2 Gene expression
Gene expression is an interpretation of data encoded in quality into protein or RNA structures that are available and working in the cell. Communicated qualities incorporate qualities that are interpreted into delivery person RNA (mRNA) and afterward converted into protein, and in addition, qualities that are deciphered into RNA, for example, exchange and ribosomal RNAs, yet not converted into protein. Proposed ESFSC, a method that simulating Random Walker for the identification of disease-related genes using topological properties of genes and gene expression correlation. The authors formulated the seed set based on known disease genes and their k-nearest neighbor, and initial probability scores were assigned to genes on the basis of the location of the gene. Furthermore, random walk with restart was implemented which was guided by similarity transition matrix and correlational transition matrix constructed on the basis of topological similarity properties and gene expression profiles, respectively. The final ranking of the genes was then done based on the steady-state probabilities obtained by combing results of random walk of two different matrices.
4.3 Orthology
Orthologs are homologous proteins that are gotten from a typical precursor. They generally have high comparable amino corrosive successions and hold the same or fundamentally the same as capacities. Peng et al. (2012) used the orthologous property of genes to perform gene prioritization. The authors proposed a method ION which combines three features: connections among the genes, the orthologous property of genes and features of neighbors. Initially, the orthologous score was being assigned to each gene and then the adjacency matrix was calculated based on the edge clustering coefficient. Then the rank of each gene is calculated on the basis of orthologous score and neighbor induced score.
4.4 Protein complexes
Yang et al. (2011) proposed a method RWPCN which perform prediction and prioritization of genes using human protein complexes and PPI network. The integrated network consisted of three layers with the phenotype network at top connecting phenotypes based on similarity scores, protein complex network connecting phenotypically-related protein complexes in the middle and PPI network connecting two interacting at the bottom. The authors have used both random walk algorithm and flow propagation for assigning scores to proteins which are further used for ranking them for prioritization. Jiang et al. (2014) proposed a random-set scoring model using protein complexes and text mining of biomedical literature for prioritizing the genes in a protein network. The authors’ collected gene-associated phenotype data from three different sources and the vocabulary filters defining phenotypic profiles from four sources and the cosine coefficient between the two represent the semantic similarity.
4.5 Tissue-specific information
Deng et al. (2016) proposed disease gene prioritization on a heterogeneous network using tissue-specific information. The heterogeneous network was built by the integration of tissue-specific PPI networks with phenotypic features. The authors implemented the proposed heterogeneous network approach on other existing prioritization approaches: Random Walk with Restart (RWR), PRINCE, Guo’s Method used individually.
Xi et al. (2016) prioritized atherosclerosis-related genes using text mining by performing the systematic analysis: pathway analysis, functional clustering analysis, and gene network analysis. The functional clustering analysis of genes that including gene ontology (GO) analysis identified over-represented genes. The pathway analysis identified the most highly enriched pathway and the gene network analysis prioritized genes following the hub method. Table 6 contains a summary of methods using the heterogeneous type of data.
4.6 Protein expression profile
Ren et al. (2019) have evaluated the performance of local ranking and global ranking methods by integrating protein expression profiles with the PPI network. Three different protein sets with prior disease knowledge are used. For local ranking methods, they ranked genes using the number of disease genes directly related to the gene and for global ranking, heat kernel method for restarting random walk on one of the three networks with prior disease knowledge is used.
Table 6 contains the summary of proposed network-based techniques using a heterogeneous type of data.
5 A basic workflow of network-based disease gene prioritization
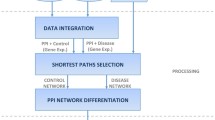
A gene prioritization problem can be formulated as given a disease (D) of interest and a list of genes, the task is to rank these genes based on the likelihood of the genes to be involved with disease. The approach to the problem using the network-based technique is shown in Fig. 1 with details of each as follows:

Network-based disease gene prioritization approach
-
Collection of candidate genes: The quality of the prioritization process is greatly affected by the set of genes that needs to be prioritized. In addition, the size of the candidate set is a major factor, i.e., it can be a group of genes or the entire genome. There are various primary and secondary data sources such as microarray data, data from public repositories, data extracted from other biomedical literature available online which can be used for experimentation purposes.
-
Collection of disease-related information: With the advancing research, the disease-related knowledge is now available abundantly which can either be extracted from the biomedical literature online or from public databases that have been specially formulated to gather the information. This disease-related information is called the seed set(S).
-
Preprocessing of data: There are different ways in which data is represented such as a matrix of numbers, graphs, list of terms, etc. The way data is represented also influence the way the prioritization technique is being used. In addition, in case of data integration data from multiple sources needs to be merged efficiently.
-
Merging of information: Next the merging of both types of data, i.e., candidate data and disease-related information is done. This is an optional step and depends on the computational strategy being implemented for prioritization.
-
Implementation of computational strategy: After the formulation and preprocessing of the data set, the computational approach for prioritization is being implemented on it. These computational approaches are of various types and have been discussed in detail in the previous sections. Thus, on the basis of various features, the process of prioritization is performed which gives as output a list of genes ranked on the basis of the probability of these genes to be involved in the disease.
-
Assessment of prioritization: Proper assessment of the system is very important to analyze the effectiveness of the system such as benchmarking, cross-validation, using the negative dataset, functional enrichment, etc. Various performance measures such as the area under ROC curves, sensitivity, specificity, fold enrichment, etc. are used to validate and compare the performance of the system with other existing systems.
6 Discussion
Disease gene prioritization aims at identifying the genes which are most likely to be related to the disease. It is helpful for doctors to identify target genes and designing efficient treatment systems. It also is helpful in the prevention and early diagnosis of disease. In this review, various approaches to network-based disease gene prioritization based on PPI networks have been discussed. Although with advancing technology various approaches have been proposed still there are problems which need to be handled.
The biological data is the core of gene prioritization models and the performance of any prioritization model highly depends on the quality of data. One of the main problems with the data is the incompleteness of the data. Although there are many techniques available for identifying interactions among genes still there are missing links in the data. These missing links limit the performance of the system. Mean or median of observed values could be one possible way to interpolate missing data. In addition, the biological data is retrieved using parsing of available literature, so there is a possibility of errors due to falsely identified genes or missing links. This degradation in the quality of data could be improved by integrating data from various sources.
Data integration from different sources is assumed to deal with the problem of missing and falsely identified data. But acquiring and integrating data from various sources itself faces the problem of lack of consistent representation of data, as the same concept can be represented in different ways which sometimes leads to redundant data. Almost all approaches work on the basis of the prior knowledge available about the disease, but due to incompleteness of data, the results are biased towards the genes previously identified to have some relation to the disease. The approaches which have training set dependent on the disease are biased towards the well-studied set of genes.
So, one way to deal with these problems is to first integrate data from different data sources and then process that data by retrieving only the data which have significance in multiple data sources and leaving those with lesser significance. Another way is to use heterogeneous data related to genes such as ontology, annotations, pathways, etc. to improve the performance of the system. However, combining data from multiple resources is also a difficult and time-consuming task.
Another problem related to gene prioritization approaches is the regular updation of data sources. With the passage of time, the datasets also keep changing. So whenever the dataset is updated, the techniques which are parameter dependent needed to be returned to optimize the performance. Since all changes in a system cannot be automated it is difficult for the researchers to keep updating the data frequently.
Many researchers have proposed approaches based on the integration of heterogeneous types of biological data into a single network. Even though these approaches performed better as compared to others but integrating and organizing biological data into layers of networks is a difficult task as all elements are interrelated. In addition, biological information about various species is not available.
Different approaches use a different type of data and format. So, there is no standard data to compare the performance of different approaches. In addition, there is a need for improvement in benchmarking efforts to provide the global platform for the comparison of approaches.
7 Conclusions and future scope
In this review, the importance of the disease gene prioritization technique has been highlighted. We discussed various categories into which the technique has been categorized. The filtering-based techniques excludes the gene even if it does not match a single criterion without considering others. The similarity-based methods often fail to handle diseases about which no prior association information is known. Thus, major focus was to point out the progress made in disease gene prioritization techniques based on the PPI network. Gene prioritization techniques use various properties of proteins and genes to rank them on the basis of their locational proximity to the disease genes. An improvement over it is to use data from multiple resources which reduces the effect of missing and false data. Integration could be done in two ways either by first combing data from heterogeneous sources and then perform ranking or by individually ranking genes from different resources and then combine the results of ranked lists into one. But the performance of these topology-based methods is reasonable as the results are biased by the network topology. Thus, the performance of prioritization techniques could be enhanced by integrating different types of biological knowledge about genes with the network information. This integration attributes to further performance enhancement of techniques as knowledge integration covers rich information that could not be captured by network analysis. This integration of information can be made more productive with the help of functional analysis. Functional clustering analysis identifies the disease-causing genes based on the annotations which are significantly enriched. This method also identifies the annotations which are overly represented. Using enrichment analysis to combine different types of information can help researchers to use annotations that are more functionally enriched which might yield better results. Higher performance enhancement can be expected when more complete datasets are used.
Availability of data and material
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
Adie EA, Adams RR, Evans KL, Porteous DJ, Pickard BS (2006) SUSPECTS: enabling fast and effective prioritization of positional candidates. Bioinformatics 22(6):773–774
Aerts S et al (2006) Gene prioritization through genomic data fusion. Nat Biotechnol 24(5):537–544
Amberger JS, Bocchini CA, Scott AF, Hamosh A (2019) OMIM.org: leveraging knowledge across phenotype–gene relationships. Nucleic Acids Res 47(1):1038–1043
Aragues R, Jaeggi D, Oliva B (2006) PIANA: protein interactions and network analysis. Bioinformatics 22(8):1015–1017
Bader GD, Betel D, Hogue CW (2003) BIND: the biomolecular interaction network database. Nucleic Acids Res 31(1):248–250
Bergmann S, Ihmels J, Barkai N (2004) Similarities and differences in genome wide expression data of six organisms. PLoS Biol 2(1):E9
Blaschke C, Andrade M, Ouzounis C, Valencia A (1999) Automatic extraction of biological information from scientific text: protein–protein interactions. In Proceedings of 1st international conference on intelligent systems for molecular biology 7:60–67
Boutros M, Ahringer J (2008) The art and design of genetic screens: RNA interference. Nat Rev Genet 9(7):554–566
Breitkreutz B, Stark C, Tyers M (2003) Osprey: a network visualization system. Genome Biol 4(3):R22
Chen J, Aronow B, Jegga A (2009) Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinform 10(1):73–87
Cline M et al (2007) Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2(10):2366–2382
Dahlquist K et al (2002) GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet 31(1):19–20
Deng Y, Gao L, Guo X, Wang B (2016) Integrating phenotypic features and tissue-specific information to prioritize disease genes. Sci China Inf Sci 59(7):1–10
Dezső Z et al (2009) Identifying disease-specific genes based on their topological significance in protein networks. BMC Syst Biol 3(1):36
Erten S, Bebek G, Koyutürk M (2011) Vavien: an algorithm for prioritizing candidate disease genes based on topological similarity of proteins in interaction networks. J Comput Biol 18(11):1561–1574
Franke L et al (2004) TEAM: a tool for the integration of expression, and linkage and association maps. Eur J Hum Genet 12(8):633–638
Funahashi A, Morohashi M, Kitano H, Tanimura N (2003) Cell Designer: a process diagram editor for gene-regulatory and biochemical networks. Biosilico 1(5):159–162
Gentili M, Martini L, Petti M, Farina L, Becchetti L (2019) Biological random walks: integrating heterogeneous data in disease gene prioritization. s.l., s.n
Ghiassian SD, Menche J, Barabási AL (2015) A DIseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome. PLoS Comput Biol 11(4):e1004120
Gonçalves J, Francisco A, Moreau Y, Madeira S (2012) Interactogeneous: disease gene prioritization using heterogeneous networks and full topology scores. PLoS ONE 7(11):e49634
Grewal N, Singh S, Chand T (2016) Effect of aggregation operators on network-based disease gene prioritization: a case study on blood disorders. IEEE/ACM Trans Comput Biol Bioinform 14(6):1276–1287
Hirschhorn J, Gajdos Z (2011) Genome-wide association studies: results from the first few years and potential implications for clinical medicine. Ann Rev Med 62:11–24
Hsu CL, Huang YH, Hsu CT, Yang UC (2011) Prioritizing disease candidate genes by a gene interconnectedness-based approach. BMC Genom 12(3):S25
Hu Z et al (2005) VisANT: data-integrating visual framework for biological networks and modules. Nucleic Acids Res 33(2):W352–W357
Indulekha T.S, Aswathy GS, Sudhakaran P (2018) A graph based algorithm for clustering and ranking proteins for identifying disease causing genes. In 2018 international conference on advances in computing, communications and informatics (ICACCI), Bangalore IEEE:1022–1026
Ito T et al (2000) Toward a protein–protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc Natl Acad Sci 97(3):1143–1147
Jiang L et al (2014) A random set scoring model for prioritization of disease candidate genes using protein complexes and data-mining of GeneRIF OMIM and PubMed records. BMC Bioinform 15(1):315–327
Jianzhen Xu, Li Y (2006) Discovering disease-genes by topological features in human. Syst Biol 22:2800–2805
Köhler S, Bauer S, Horn D, Robinson P (2008) Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet 82(4):949–958
Krauthammer M, Kaufmann AC, Gilliam TC, Rzhetsky A (2004) Molecular triangulation: bridging linkage and molecular-network information for identifying candidate genes in Alzheimer’s disease. Proc Natl Acad Sci USA 101(42):15148–15153
Lage K et al (2007) A human phenome–interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol 25(3):309–316
Le D, Kwon Y (2013) Neighbor-favoring weight reinforcement to improve random walk-based disease gene prioritization. Comput Biol Chem 44:1–8
Li Y, Li J (2012) Disease gene identification by random walk on multigraphs merging heterogeneous genomic and phenotype data. BMC Bioinform 13(7):1–12
Li Y, Patra JC (2010) Integration of multiple data sources to prioritize candidate genes using discounted rating system. BMC Bioinform 11:S20
Li M et al (2014) Prioritization of orphan disease-causing genes using topological feature and GO similarity between proteins in interaction networks. Sci China Life Sci 57(11):1064–1071
Li M, Lu Y, Wang J, Wu FX, Pan Y (2015) A topology potential-based method for identifying essential proteins from PPI networks. IEEE/ACM Trans Comput Biol Bioinform (TCBB) 12(2):372–383
Licata L et al (2012) MINT, the molecular interaction database: 2012 update. Nucleic Acids Res 40(D1):D857–D861
Lim J et al (2006) A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 125(4):801–814
Liu Y, Tong H, Lei X, Tang Y (2016) Network based models and path based features for gene prioritization In 2016 IEEE 20th International Conference on Computer Supported Cooperative Work in Design (CSCWD) IEEE:129–132
Liu R, Mancuso CA, Yannakopoulos A, Johnson KA, Krishnan A (2020) Supervised-learning is an accurate method for network-based gene classification. Bioinformatics 36(11):3457–3465
Lowe HJ, Barnett GO (1994) Understanding and using the medical subject headings (MeSH) vocabulary to perform literature searches. Jama 271(14):1103–1108
Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, Kawaler E (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534(7605):55–62
Morris J et al (2014) Affinity purification–mass spectrometry and network analysis to understand protein–protein interactions. Nat Protoc 9(11):2539–2554
Morris JH, Kuchinsky A, Pico A (2015a) Analysis and visualization of biological networks with Cytoscape
Newman M, Girvan M (2004) Finding and evaluating community structure in networks. Phys Rev E 69(2):026113
Oti M, Brunner H (2007) The modular nature of genetic diseases. Clin Genet 71(1):1–11
Oti M, Snel B, Huynen M, Brunner H (2006) Predicting disease genes using protein–protein interactions. J Med Genet 43:691–698
Ott J, Kamatani Y, Lathrop M (2011) Family-based designs for genome-wide association studies. Nat Rev Genet 12(7):465–474
Peng W et al (2012) Iteration method for predicting essential proteins based on orthology and protein–protein interaction networks. BMC Syst Biol 6(87):1–17
Perez-Iratxeta C, Bork P, Andrade MA (2002) Association of genes to genetically inherited diseases using data mining. Nat Genet 31(3):319–346
Pesquita C, Faria D, Falcao AO, Lord P, Couto FM (2009) Semantic similarity in biomedical ontologies. PLoS Comput Biol 5(7):e1000443
Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D (1997) GeneCards: integrating information about genes, proteins and diseases. Trends Genet 13(4):163
Ren J, Shang L, Wang Q, Li J (2019) Ranking Cancer Proteins by Integrating PPI Network and Protein Expression Profiles BioMed Research International 2019:3907195. https://doi.org/10.1155/2019/3907195
Safari-Alighiarloo N, Taghizadeh M, Rezaei-Tavirani M, Goliaei B, Peyvandi AA (2014) Protein–protein interaction networks (PPI) and complex diseases. Gastroenterol Hepatol Bed Bench 7(1):17–31
Schlicker A, Lengauer T, Albrecht M (2010) Improving disease gene prioritization using the semantic similarity of Gene Ontology terms. Bioinformatics 26(18):i561–i567
Seelow D, Schwarz JM, Schuelke M (2008) GeneDistiller—distilling candidate genes from linkage intervals. PLoS ONE 3(12):e3874
Shannon P et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504
Sinsha K, Chitturi B (2016) A study of gene prioritization algorithms on PPI. In- International conference on advances in computing, communications and informatics (ICACCI), Jaipur IEEE:927–934
Stelzl U et al (2005) A human protein–protein interaction network: a resource for annotating the proteome. Cell 122(6):957–968
Stuart JM, Segal E, Koller D, Kim SK (2003) A gene-coexpression network for global discovery of conserved genetic modules. Science 302(5643):249–255
Szklarczyk D et al (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39(1):D561–D568
Van Driel MA, Bruggeman J, Vriend G, Brunner HG, Leunissen JA (2006) A text-mining analysis of the human phenome. Eur J Hum Genet 14(5):535–542
Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R (2010) Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol 6(1):e1000641
Wang J, Chen G, Li M, Pan Y (2011) Integration of breast cancer gene signatures based on graph centrality. BMC Syst Biol 5(3):S10
Weckwerth W, Loureiro M, Wenzel K, Fiehn O (2004) Differential metabolic networks unravel the effects of silent plant phenotypes. Proc Natl Acad Sci USA 101(20):7809–7814
Wood L et al (2007) The genomic landscapes of human breast and colorectal cancers. Science 318(5853):1108–1113
Xenarios I et al (2002) DIP, the database of Interacting proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res 30(1):303–305
Xi D, Zhao J, Lai W, Guo Z (2016) Systematic analysis of the molecular mechanism underlying atherosclerosis using a text mining approach. Hum Genom 10(1):1–8
Yang P et al (2011) Inferring gene–phenotype associations via global protein complex network propagation. PLoS ONE 6(7):e21502
Yu S et al (2008) Comparison of vocabularies, representations and ranking algorithms for gene prioritization by text mining. Bioinformatics 24(16):i119–i125
Yu S, Tranchevent LC, De Moor B, Moreau Y (2011) Kernel-based data fusion for machine learning: methods and applications in bioinformatics and text mining, 1st edn. Springer, Berlin
Zhang W, Sun F, Jiang R (2011) Integrating multiple protein–protein interaction networks to prioritize disease genes: a Bayesian regression approach. BMC Bioinform 12(Suppl 1):S11
Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, Davies SR (2014a) Proteogenomic characterization of human colon and rectal cancer. Nature 513(7518):382–387
Zhang SW, Shao DD, Zhang SY, Wang YB (2014b) Prioritization of candidate disease genes by enlarging the seed set and fusing information of the network topology and gene expression. Mol BioSyst 10(6):1400–1408
Zhang W, Lei X, Bian C (2019) Identifying Cancer genes by combining two-rounds RWR based on multiple biological data. BMC Bioinform 20(Suppl 18):518
Zhu C, Kushwaha A, Berman K, Jegga AG (2012) A vertex similarity-based framework to discover and rank orphan disease-related genes. BMC Syst Biol 6(3):1–9
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Code availability
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaushal, P., Singh, S. Network-based disease gene prioritization based on Protein–Protein Interaction Networks. Netw Model Anal Health Inform Bioinforma 9, 55 (2020). https://doi.org/10.1007/s13721-020-00260-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-020-00260-9