
Abstract
Increasing energy expenditure is an appealing therapeutic target for the prevention and reversal of metabolic conditions such as obesity or type 2 diabetes. However, not enough research has investigated how to exploit pre-existing neural pathways, both in the central nervous system (CNS) and peripheral nervous system (PNS), in order to meet these needs. Here, we review several research areas in this field, including centrally acting pathways known to drive the activation of sympathetic nerves that can increase lipolysis and browning in white adipose tissue (WAT) or increase thermogenesis in brown adipose tissue (BAT), as well as other central and peripheral pathways able to increase energy expenditure of these tissues. In addition, we describe new work investigating the family of transient receptor potential (TRP) channels on metabolically important sensory nerves, as well as the role of the vagus nerve in regulating energy balance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Obesity is one of the fastest growing non-communicable diseases worldwide [1], and recently, the alarming statistics have been updated to include even greater numbers of affected individuals in the USA. CDC data from 2013, estimating 69 million obese people in the USA alone, appears to be short by 12 million people [2], bringing the actual total closer to 81 million. Obesity is characterized by the accumulation of adipose mass due to energy intake exceeding energy expenditure and is a chronic disorder of energy imbalance that is a major risk factor in the pathogenesis of many diseases. Obese individuals are at risk for developing a wide range of mechanical and physiological disorders including the following: sleep apnea, osteoarthritis, cardiovascular diseases, and type 2 diabetes. While diet-induced obesity (DIO) can be reversible, many of its comorbidities are not. Therefore, understanding the mechanisms involved in the control of energy balance can help tip the scales in favor of energy expenditure when disequilibrium occurs, thus alleviating deleterious obesity-related health effects (Fig. 1).

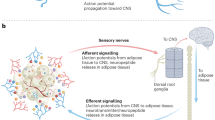
Central and peripheral drivers of energy expenditure in BAT and WAT. Sensory neurons convey environmental information to the central nervous system (CNS) which responds accordingly to regulate energy balance by relaying information to peripheral tissues via the sympathetic nervous system (SNS). Vagal efferent pathways can act in different ways to increase energy expenditure (E.E.), while vagal nerve stimulation prompts the CNS to decrease food intake and Increases expression of neutrophic factors which may also help to stimulate E.E. Certain vagal nuclear receptors are involved in peripheral lipid sensing, another way to potentially regulate E.E. The nociceptive TRP channels are found on sensory neurons and expressed in a variety of tissues including the skin and adipose. Agonism of certain TRP channels can drive increased sympathetic output via action on the CNS. When acting directly on adipose tissue, TRP channel agonism has been shown to inhibit adipogenesis in white and brown adipocytes. However, conflicting evidence remains, as studies show disparaging phenotypes with the same TRP channel knockout murine models. Circulating factors, such as the adipose-secreted hormone leptin, act in the CNS to increase E.E. via hypothalamic pathways. Other circulating factors such as FGF21 can act not only on the CNS to increase sympathetic tone (via the suprachiasmatic nucleus of the hypothalamus or the dorsal vagal complex of the hindbrain) but also directly on adipose tissue to stimulate energy-expending processes such as lipolysis and thermogenesis
Energy balance involves a complex network of central nervous system (CNS) communication with the periphery, including bidirectional neural communication with adipose tissues as well as CNS detection of secreted endocrine products from adipose tissues, termed adipokines. White adipose tissue (WAT) adipocytes store energy as lipid droplets, a significant evolutionary adaptation, and adipocytes in brown adipose tissue (BAT) have the ability to use lipids for fuel through the process of non-shivering thermogenesis, which can defend against obesity and diabetes [3]. Indeed, when one considers the two arms of energy balance, energy intake and energy expenditure, aspects of energy expenditure are largely regulated by the CNS, WAT, and BAT, making the study of these tissues very important. Aspects of energy expenditure regulated by these tissues include the following: physical activity and exercise (controlled by CNS motivation pathways and neural communication with muscle), basal metabolic rate (controlled by tissue mitochondria, including mitochondria-rich tissues such as BAT), and thermogenesis (largely controlled by UCP1 in BAT).
Circulating endocrine factors, secreted either from adipose tissue or other metabolic tissues such as liver or muscle, as well as the CNS regulation of sympathetic nerves that can activate lipolysis in WAT or thermogenesis in BAT, are important research topics in the regulation of whole-body energy balance. Catecholamine neurotransmitters such as norepinephrine, which are released by nerve terminals in the sympathetic nervous system (SNS), are a positive regulator of lipolysis. In addition, sympathetic activation of WAT, such as with cold exposure, can stimulate the formation of brown adipocytes in that depot, through a process called “browning.” These recruitable/inducible brown adipocytes also may contribute to thermogenesis and energy expenditure, and the identification of CNS or endocrine pathways mediating the development and activation of these brown adipocytes is an active area of research for increasing energy expenditure to combat metabolic diseases. In addition, increasing the mass or activation of brown adipocytes in the body may also help alleviate some of the comorbidities of obesity, including insulin resistance and diabetes [4].
Here, we review the current understanding of several mechanisms involved in central and peripheral neural activation of energy expenditure in both white and brown adipose tissues, including the regulation of lipolysis and thermogenesis, and we present emerging molecular targets for potentially increasing energy expenditure to attenuate obesity in humans.
Neural Circuitry Between Adipose Depots and the Brain
The use of the retrograde, sympathetic-specific transneuronal viral tract tracer pseudorabies virus (PRV152) has shown SNS outflow to BAT from specific regions of the CNS [5], while the use of an anterograde sensory-specific transneuronal viral tract tracer (H129 strain of herpes simplex virus −1) has demonstrated the sensory system feeding back from BAT to the brain [6]. By combining the retrograde PRV152 with anterograde H129 tracers, the latest work in transneuronal tracing suggests that there is crosstalk between the SNS and sensory neural innervation of BAT and that the SNS-sensory feedback loop contains a highly coordinated and redundant neuronal network [7]. Of particular interest is the periaqueductal gray (PAG) area of the midbrain where SNS-sensory circuits innervating BAT overlap. The PAG is also an area rich in oxytocin receptors, which have been shown to regulate thermogenesis (see below). It now appears that differential SNS nodes within the midbrain are responsible for outflow to different fat pads [8], indicating that SNS activation of BAT could be accomplished by targeting specific pathways regulating drive to that depot, without affecting sympathetic activity in other tissues that may lead to undesirable effects.
Similar transneuronal tracing experiments have been undertaken in WAT. Early studies used fluorescent tract tracers (i.e., DiI, FlouroGold) to demonstrate bidirectional projections of postganglionic neurons residing in the sympathetic chain to WAT [9]. These studies suggested neuronal segregation dependent on the WAT pad being innervated [10], a suspicion that was verified many years later through the use of PRV tracers. Injection of distinct viral fluorescent reporters into mesenteric and subcutaneous WAT revealed segregation of neurons innervating these WAT pads [11]. This segregation may partially explain why certain WAT depots show differential mobilization of lipids or browning patterns in response to sympathetic stimulation. For a more complete description of the CNS-SNS-WAT circuitries, see reviews [8, 9].
Anterograde tracing applied to intrascapular WAT of rats revealed unsurprisingly that the tissue also possesses sensory innervation, evidenced by labeling of neurons in the dorsal root ganglia [12]. This sensory feedback loop allows for ascending neural communication between WAT and CNS. Therefore, both BAT and WAT are in direct communication with the CNS in a bidirectional manner via both sensory and sympathetic nerves.
Vagal Efferent Pathways Regulating Energy Expenditure
Cold stimulation is known to activate BAT depots and initiate non-shivering thermogenesis; however, cold acclimation as a means of increasing energy expenditure is not necessarily the most attractive obesity therapy given the potential for discomfort and compensatory increases in appetite. There does exist the potential to directly stimulate the neural pathways that mimic cold-induced energy expenditure without the cold exposure, and the vagus nerve may represent such a target.
Vagal sensory neurons in the nodose ganglion (NG) innervate visceral organs such as the GI tract, pancreas, liver, and portal vein, thus linking peripheral metabolic cues (i.e., circulating fuels) with the CNS and providing information on satiety in order to help control feeding behavior. Clinical observations of weight loss associated with long-term vagus nerve stimulation (VNS) as a treatment for epilepsy [13, 14] and severe depression [15] have led to further investigation of the role of vagal nerves in metabolism. Subsequent rodent studies revealed that long-term VNS led to resistance to DIO, with a concurrent reduction of epididymal fat mass. This was conferred by reduced food intake due to increased satiety signaling to the CNS [16]. Further investigation revealed that VNS increased the expression of neurotrophic factors, including brain-derived neurotrophic factor (BDNF), as well as increased norepinephrine concentration in the brain of rats [17], which may also play a role in stimulating peripheral energy expenditure [18].
A mechanism for these observations remained unclear, and the involvement of vagal-induced BAT activation was conflicting. Some studies showed no effect of a cervical vagotomy on BAT activity [19], while others reported a decrease in both WAT and BAT weight after a bilateral subdiaphragmatic vagotomy in obese rats [20]. Still, others demonstrated that truncal vagotomy prevented the inhibitory effects of ghrelin on norepinephrine release in BAT [21], thereby suggesting a link between vagal signaling and BAT thermogenesis.
More recently, Lui et al. elucidated a potential mechanism by which vagal nerves help regulate energy balance in a murine model. Their study revealed that vagal neurons express several nuclear receptors, including PPARγ, which can directly interact with diet-derived lipids. After determining that a high-fat diet (HFD) reduces expression of PPARγ in the NG, they demonstrated that deletion of PPARγ in Phox2b neurons, which allows for ablation of PPARγ function in both central and peripheral regions of vagal afferents, resulted in a pronounced reduction of fat mass without a reduction in food intake while on a HFD [22•] which was indicative of an increase in energy expenditure. Along these lines, they were also able to demonstrate that deletion of PPARγ in Phox2b neurons promoted thermogenesis and browning of WAT [22•]. This study revealed that PPARγ in NG neurons interacts with dietary-derived lipids, and although the physical mechanisms of this interaction remain to be resolved, it is clear that PPARγ in this context regulates gene expression related to lipid metabolism, as evidenced by the downregulation of CD36, lipoprotein lipase, and aP2 in PPARγ-deficient NG neurons [22•]. These findings suggest a dual role for PPARγ in vagal neurons, whereby under normal dietary conditions, it acts to suppress energy expenditure, yet with HFD downregulation of PPARγ in vagal sensory neurons, it appears to promote diet-induced thermogenesis in order to compensate for the increase in adiposity, at least in the short-term [22•]. Lui et al. observed strongly increased UCP1, Adrβ3, and other browning markers in subcutaneous WAT despite a modest, albeit significant, increase in UCP1 expression in BAT, which may indicate that browning of WAT via increased sympathetic tone is the driving force behind the energy expenditure in this model.
The same group then went on to show that loss of another nuclear receptor, liver X receptor alpha and beta (LXRα/β), in the NG of the vagus nerve also led to increased energy expenditure, resistance to DIO, browning of WAT, and an increase in skeletal muscle mitochondrial respiration [23]. LXRs are oxysterol-sensing NRs, involved in cholesterol uptake, transport, and excretion in various tissues, including the liver and nervous system [23]. Similar to their previous study of PPARγ deletion from vagal afferents, loss of LXRα/β in a subset of vagal sensory neurons (those expressing the Nav1.8 ion channel) led to a more pronounced effect on energy expenditure and weight loss when mice were on HFD, suggesting that LXRs may regulate energy expenditure based on the availability of lipid or cholesterol [23].
TRP Channels and Sensory Nerve Integration
Transient receptor potential (TRP) channels are Ca2+ permeable non-selective cation channels found on sensory neurons and have been extensively studied for their role in pain sensation. They are a group of diverse cellular sensors for an array of physical and chemical stimuli, such as sensations of touch, taste, temperature, and pain [24]. Although they play a crucial role in a variety of cell signaling processes sending sensory transduction, the molecular mechanisms involved in TRP channel activation differ depending on the type of TRP channel—many exhibiting polymodal activations via voltage, temperature, ligand binding, or mechanical force [24]. Most TRP channels are also chemoreceptors for various naturally occurring substances, including capsaicin, which can be found in edible plants. Capsaicin mimics the effects of cold exposure by agonizing transient receptor potential vanilloid subtype 1 (TRPV1), which leads to recruitment of BAT, increasing energy expenditure and decreased adiposity [25]. TRPV1 was first identified in dorsal root ganglion (DRG) and trigeminal ganglion (TG) neurons and is also highly expressed in spinal and peripheral nerve terminals and several non-neuronal tissues including skin, muscle, gut, [24, 26], pancreas, and visceral adipose tissue [27]. In the pancreas, it is involved in substance P (SP) release during pancreatitis [28]. The majority of TRPV1-positive neurons respond to nerve growth factor (NGF) and express calcitonin gene-related peptide (CGRP) and SP [26]. Zhang et al. detected TRPV1 channels in 3T3-L1 cells and found that capsaicin inhibits adipogenesis in these cells, an effect that can be reversed by RNA-interference knockdown of TRPV1 [27]. Furthermore, long-term supplementation of capsaicin to HFD prevented obesity in WT but not TRPV1 knock-out (KO) mice [27].
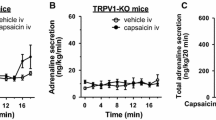
In a contradictory study, whole body TRPV1 KO mice showed resistance to DIO without a decrease in food intake, when compared to littermate controls [29]. Albeit the diets used in the study were conservative in fat content (4.5 vs 11 % HFD); nevertheless, increased energy expenditure in the TRPV1-null mice was observed and implicated as the cause for weight disparity. The study also revealed that 3T3-L1 cells are sensitive to CGRP but not SP, both products released by TRPV1 sensory neurons [29]. Since CGRP has been reported to promote neurogenesis [30], and the authors show a functional response to CGRP in 3T3-L1 cells, they propose that TRPV1 regulates adipocyte function through a neurogenic pathway. These findings in the TRPV1-null contradict the previously described role of TRPV1 in obesity and warrant further investigation.
In recent rodent studies, topical application of capsaicin decreased visceral WAT depot size along with reduced lipid droplet size, together indicative of fuel mobilization via lipolysis. This also was found to reduce insulin resistance in DIO mice [31]. Ohyama et al. recently demonstrated a combination of exercise and capsinoid supplementation has an additive effect on increasing energy expenditure in mice, resulting in an overall protective effect against DIO [32]. Capsinoids are non-pungent analogs of capsaicin and are being studied as a treatment for obesity due to their ability to modulate SNS activity and increase energy expenditure via increased TRPV1 channel activation [33]. TRPV1 agonists are known to increase sympathetic tone via activation of the CNS [33]. Based on evidence that TRPV1 acts to regulate body temperature in various species [34], Alawi et al. hypothesized that TRPV1 may act tonically suppress the SNS. They found that TRPV1 antagonism resulted in a state of hyperthermia in WT mice, accompanied by increased norepinephrine in BAT, but not in TRPV1 KO mice [35•]. However, TRPV1 KO mice have normal body temperature under basal conditions indicating a developmental compensatory effect [35•]. Indeed, TRPV1 KO mice exhibit a reduction in capacity to respond to the sympathomimetic drug d-amphetamine, suggesting that sympathetic drive has been reduced in these animals, which may have normalized body temperature [35•].
Dietary capsaicin, which leads to TRPV1 activation, can also increase energy metabolism in skeletal muscle of wild type mice. Chronic TRPV1 activation induces PGC-1α and increases mitochondrial biogenesis in a calcium channel-dependent manner [36], effects which were absent in TRPV1 null mice [36]. PGC-1α is a well-known regulator of lipid and glucose metabolism, mitochondrial biogenesis, and muscle fiber development and plays a crucial role in the activation of genes involved in the browning of WAT. These data provide a mechanism by which TRPV1 activation increases energy expenditure.
Several studies revealed that activation of certain TRP channels within the vanilloid family could inhibit adipogenesis. TRPV3 activation with chemical agonists (such as diphenylborinic anhydride (DPBA), a synthetic non-selective TRP channel agonist; and epicatechin, a compound found in cocoa and green tea) prevents lipid accumulation and adipogenesis in 3T3-L1 preadipocytes, by downregulating the PI3K/Akt pathway in a dose-dependent manner, without altering the expression of TRPV3 [37•]. Increased activation of TRPV3 prevents forkhead box protein O1 (FOXO1) phosphorylation (FOXO1 is a mediator of adipogenesis), which in turn downregulates the adipogenic genes C/EBPα and PPARγ [37•]. Furthermore, mice supplemented with either of the TRPV3 agonists (DPBA or epicatechin) may have been protected from HFD-induced obesity, as evidenced by fat pad size comparable to chow-fed mice [37•]. Since food intake is not altered by TRPV3 agonism, an increase in energy expenditure could have been observed to explain these findings; however, no such measure was presented. Interestingly, visceral and subcutaneous adipose tissue have lower TRPV3 expression in mice fed a HFD as well as ob/ob mice and db/m + mice, when compared to chow-fed or WT mice, respectively [37•], indicating a role in the obese phenotype.
In a recently published study, Sun et al. found that TRPV2 is highly and functionally expressed in mouse brown adipocytes and intrascapular BAT [38•]. Like other TRP channels within the vanilloid family, TRPV2 possesses polymodal activation mechanisms, including activation via temperature and chemical ligands [24, 34]; however, TRPV2 at >53 °C has the highest temperature threshold for activation [24, 34]. Similarly, TRPV1 and TRPV3 play a role in white adipogenesis [27, 37•], while TRPV2 is involved in brown adipogenesis [38•]. In in vitro studies, pharmacological agonism of TRPV2 prevented mouse brown adipocyte differentiation [38•]. In TRPV2 KO cells, a higher differentiation cell count and triglyceride level was observed, compared to wild type, but only when the differentiation medium was diluted ten times [38•]. The authors explain this discrepancy with the notion that under control conditions, differentiation became saturated, blunting any significant finding. This suggests that a secondary stressor might play a role in TRPV2-mediated brown adipocyte differentiation inhibition.
While wild type brown adipocytes showed a robust response to pharmacological β-adrenergic stimulation evidenced by an increase in thermogenic markers UCP1 and Pgc1α, the TRPV2 KO cells did not. Furthermore, TRPV2-dependent induction of thermogenic genes appears to involve a cyclic adenosine monophosphate (cAMP) pathway [38•]. Moreover, TRPV2 KO mice exhibited impaired thermogenic function when cold stimulated, concomitant with a decrease in core body temperature, an overall increase in adiposity, and an accumulation of large lipid droplets in BAT to the point of apparent “whitening” [38•]. Loss of TRPV2 also exacerbated an obese phonotype and insulin resistance but only on a high-fat diet. Although TRPV2 KO mice show clear impairment of BAT thermogenic function, the WAT depots appeared to not be significantly affected [38•]. This would suggest that while TRPV2 plays an essential role in the induction of a thermogenic program in BAT, its role in WAT is negligible at best, and this may explain why TRPV2 KO mice did not exhibit insulin resistance until challenged with a HFD. Browning of WAT via TRPV2-independent mechanisms may be enough to attenuate some of the energy imbalance; however, this compensation was not enough to withstand a HFD. Since TRPV2 activation increases intracellular calcium concentrations, this may be promoting thermogenic gene induction after β-adrenergic receptor activation [38•] and could explain why the TRPV2 KO mice showed impaired thermogenesis, while cell autonomous effects of the TRPV2 KO could explain the observed increase in brown adipocyte differentiation.
CNS Activation of BAT and WAT Energy Expenditure
Activation of BAT thermogenesis is accomplished through a core thermoregulatory network in the CNS. The forebrain, hypothalamus, and brainstem receive information regarding energy status, including from peripheral molecules such as insulin, leptin, and thyroid hormones. The CNS also receives incoming sensory nerve feedback, as described above. Detailed CNS regulation of thermogenesis has been extensively detailed by others [39, 40], and the aim here is to give a general overview of the circuitry involved. This neural network begins with afferent nerves delivering temperature information from warm- and cold-sensing skin receptors (i.e., TRP channels). These then excite primary sensory neurons in the dorsal root ganglia (DRG), which relay thermal information to second-order neurons in the dorsal horn (DH) [40]. Cool sensory neurons in the DH release glutamate, which activates third-order sensory neurons of the lateral parabrachial nucleus, while warm sensory DH neurons project to third-order sensory neurons in the dorsal subnucleus of the lateral parabrachial nucleus [40]. From here, the thermoregulatory response is transmitted to the preoptic area (POA) in the hypothalamus, which then produces efferent output signals delivered to thermogenic tissues via the sympathetic nervous system (SNS) [39, 40]. Preoptic warm-sensitive neurons in the POA, which are excited by glutamatergic inputs from warm-activated neurons in the median preoptic nucleus, regulate BAT thermogenesis by inhibiting sympathoexcitatory neurons in the dorsomedial hypothalamus and dorsal hypothalamic area under warm conditions [40]. During cold exposure, this inhibition is reversed and excites BAT sympathetic premotor neurons in the rostral ventromedial medulla, some of which can release glutamate thereby further exciting sympathetic preganglionic neurons and increasing BAT sympathetic nerve activity [40]. Some of the CNS-mediated signaling pathways known to affect sympathetic activation of energy expenditure are outlined below.
Leptin is a hormone exclusively synthesized by adipose tissue, which acts on the hypothalamus to regulate energy balance by promoting satiety and increasing sympathetic activation of BAT and lipolysis of WAT. As recently reviewed by [41], leptin acts centrally and peripherally in its regulation of energy balance. Briefly, leptin reaches the CNS by crossing the blood–brain barrier where binding with leptin receptor (Ob-Rb) expressed in hypothalamic regions including the arcuate nucleus (ARC) activates the JAK2/STAT3 pathway to regulate the synthesis of different neuropeptides involved in the regulation of food intake and energy balance [41]. Subsets of neurons in the ARC respond to leptin and promote either a positive or negative energy balance. For example, neuropeptide-Y (NPY) and agouti-related peptide (AgRP) expressing neurons of the ARC stimulate food intake and inhibit energy expenditure, and both of these can be inhibited by leptin [42]. Nearby pro-opiomelanocortin (POMC) neurons are stimulated by leptin and release the anorectic peptide α-melanocyte-stimulating hormone (α-MSH), which acts on melanocortin receptors to decrease food intake and increase energy expenditure [41, 42]. Meanwhile, in other areas of the hypothalamus, leptin acts to directly increase energy expenditure; leptin acting in the ventromedial hypothalamus (VMH) results in SNS activation evidenced by increased catecholamine secretion (norepinephrine and epinephrine) [43].
Diet-induced obesity (DIO) is marked by central leptin resistance. Yet, this resistance may be selective and not always result in decreased sympathetic outflow. Indeed, diet-induced obese and ob/ob mice may show increased sympathetic nerve activity in response to leptin [44]. This action appears to be mediated by the dorsomedial hypothalamus (DMH), as inhibition of leptin receptor function in this area resulted in attenuation of the thermogenic effects of leptin [44]. Interestingly, MC4R KO mice (lacking a functional melanocortin pathway) in a hyperleptinemic state have increased sympathetic nerve activity in BAT, demonstrating that this response happens via a melanocortin-independent pathway [44].
Leptin receptors are also expressed in peripheral tissue and peripheral sympathetic neurons [43]. With the knowledge that centrally acting leptin promotes lipolysis, and the recent finding that leptin stimulates axonal growth of sympathetic neuron in vitro, [45•] the question remained whether sympathetic nerves directly synapse on WAT. In a recent study, Zeng et al. presented evidence of direct activation of sympathetic input in WAT through a series of elegant experiments. Via optical projection tomography (with an adipose tissue clearing protocol similar to CLARITY) and multi-photon microscopy, they were able to visualize and quantify synaptic nerves that make direct contact with adipocytes. Although a small percent (8 %) of adipocytes were shown to make direct synaptic contact, this portion was enough to stimulate leptin-induced lipolysis and decrease subcutaneous fat pad mass upon local sympathetic simulation [46•]. They also presented evidence that while β-adrenergic receptors account for about 50 % of leptin-mediated lipolysis via activation of incoming sympathetic nerves, other adipocyte receptors are also involved [46•]. Together, their findings provide an alternative approach to overcoming CNS leptin resistance and peripheral mobilization of free fatty acids from subcutaneous depots, as are associated with DIO.
Bone morphogenetic proteins (BMPs) are growth factors belonging to the transforming growth factor (TGF) β superfamily and are known to have direct effects on brown and white adipose tissue development and function [47–49]. Recently, BMPs have also been implicated in CNS control of appetite regulation and may have a CNS-mediated effect on energy expenditure. Indeed, systemic treatment of obese mice with BMP7 leads to both a reduction of food intake and an increase in energy expenditure [49]. Similarly, Whittle et al. have shown that BMP8 is expressed in the hypothalamus of mice, and central administration of BMP8 increased neuronal activity in the lateral hypothalamic area (LHA) and VMH [50], both areas involved in CNS BAT activation.
Another centrally mediated pathway that can increase energy expenditure and thermogenesis is oxytocin (OT). OT and the oxytocin receptor (OXTR) are also likely involved in the CNS regulation of energy balance. In the CNS, OT is synthesized in the supraoptic nucleus and paraventricular hypothalamus (PVH), and neurons from these regions project to the pituitary gland where OT is secreted into systemic circulation. Hypothalamic OT neurons also project to brainstem regions that regulate sympathetic outflow to peripheral tissues. OT has metabolic roles regulating appetite and energy expenditure, including the regulation of BAT thermogenesis. For example, central administration of OT has been shown to induce hyperthermia [51], and OT-sensitive neurons in the lateral hypothalamus have been shown to project polysynaptically to BAT [52]. OT and OXTR knock-out (KO) mice fail to maintain proper body temperature, have abnormal BAT morphology, and develop obesity. Replacement of OXTR to the hypothalamus of the KO mice was enough to recover the thermogenesis defect in this model [53]. Moreover, both OT and OXTR knockout mice fail to maintain their body temperature and have malformed BAT, possibly lending to the development of late-onset obesity in these mice [54].
Fibroblast growth factor (FGF) 21 not only acts directly on BAT to stimulate glucose uptake and mobilize oxidative substrates [56] but it also acts centrally to stimulate BAT sympathetic nerve activity [55•]. More recently, FGF21 has been shown to cross the blood–brain barrier [56] where it produces a rather weak hypothalamic response [57]. Nonetheless, the whole hypothalamus has been implicated as the target of action in experiments showing increased energy expenditure in rats after i.c.v. administration of FGF21 [58]. FGF21 has also been detected in human cerebrospinal fluid [59] suggesting it may act endogenously on CNS FGF receptors. In addition to the energy expending effects of centrally acting FGF21, it has been shown to induce an adaptive starvation response [60]. Mediated by β-Klotho (an obligatory co-receptor of FGF21) expression in the suprachiasmatic nucleus (SCN) of the hypothalamus and the dorsal vagal complex (DVC) of the hindbrain, FGF21 increases systemic glucocorticoid levels, suppresses physical activity, and alters circadian behavior [60].
Neural Innervation and Activation of WAT
Although it is accepted that the SNS innervates adipose tissue, the idea that the parasympathetic nervous system (PSNS) does as well is fairly controversial. Some studies have presented apparent evidence of PSNS innervation to intra-abdominal adipose, suggesting this input could stimulate glucose uptake and free fatty acid update and directly influence the metabolic state of adipose tissue [61]. Others have challenged this notion with subsequent studies, showing no evidence of PSNS input to adipose tissue via immunohistochemistry [62], while there is supporting evidence for SNS input to white adipose tissue using retrograde tracers (as reviewed by [8]).
In 2009, Ruschke et al. presented some compelling evidence that proper peripheral nerve function is essential for maintaining healthy WAT adipose function. In a mouse model lacking the transcription factor basic domain helix-loop-helix protein neurological stem cell leukemia 2 (Nscl-2), which is expressed in early neurogenesis of central and peripheral nerves, the animals displayed reduced innervation and less vasculature in WAT, which was implicated as a cause of the observed obese phonotype [63]. Related to this, the Nscl-2 knockout mice not only had decreased peripheral innervation but they also appeared to have impaired adipocyte differentiation which led to accumulation of cells at the preadipocyte stage, suggesting a role for peripheral nerves in proper WAT development and function [63].
Despite these recent findings, the complex role of peripheral nerves in adipose tissue function, including energy expenditure, remains largely unexplored and warrants further investigation.
Neural Innervation and Activation of BAT
It has been well established in rodent models that non-shivering thermogenesis in BAT is directly controlled by SNS innervation and activation. Release of catecholamine neurotransmitters such as norepinephrine results in sympathetic activation of BAT via binding to cell surface β3-adrenergic receptors that in turn initiate a signaling paradigm that results in increased expression and activation of thermogenesis by mitochondrial UCP1. Cold exposure also functionally increases the activation of BAT through stimulation of noradrenergic nerve branching of two types of neurons: thin unmyelinated, and thick myelinated or unmyelinated nerves. Thin sympathetic neurons highly express tyrosine hydroxylase (TH), the rate-limiting step in the synthesis of norepinephrine [64]. NPY co-localizes with TH within the perivascular nerve network of these noradrenergic neurons. By contrast, fibers of the thick nerve types were shown to be CGRP and SP immunoreactive, expression that increased upon cold stimulation [64]. NPY is a potent orexigenic hypothalamic neuropeptide (NPY neurons are concentrated in the ARC and DMH) that can also be secreted by the SNS. Although peripheral NPY has no effect on classical brown adipogenesis [65], it can decrease energy expenditure in WAT by promoting adipogenesis and lipid accumulation while inhibiting lipolysis [66]. Since NPY is predominately active in the CNS but coexists with norepinephrine in the SNS, it may serve as a modulator of the negative feedback loop between CNS and SNS in mediating adipose energy expenditure.
Conclusions
While much research emphasis has been on the cell autonomous, endocrine, or paracrine signaling mechanisms that lead to increased lipolysis, thermogenesis, or browning (all contributors to whole-body energy expenditure), very little research has taken a “deep-dive” into the mechanisms regulating the neural control of these energetic pathways. For example, very little is known about the peripheral nerve products (i.e., neurotransmitters and neuropeptides) released from peripheral nerves and acting in WAT or BAT, as well as the role of neurotrophic factors and nerve branching or synaptogenesis in controlling energy expenditure in these tissues. In this review, we have outlined some of the exciting and key research areas currently exploring central and peripheral neural mechanisms for energy expenditure in white and brown adipose tissues, (Fig. 1) and we predict that this field of research will continue to burgeon and provide additional options for therapeutic targets to tip the scales from energy intake to energy expenditure, thereby combating obesity, diabetes, and other debilitating metabolic conditions that are currently driving up healthcare costs and lowering quality of life for affected individuals.
Abbreviations
- Adrβ3:
-
Adrenergic receptor β3
- AgRP:
-
Agouti-related peptide
- ARC:
-
Arcuate nucleus
- BAT:
-
Brown adipose tissue
- BDNF:
-
Brain-derived neurotrophic factor
- BMP:
-
Bone morphogenetic protein
- CGRP:
-
Calcitonin gene-related peptide
- CNS:
-
Central nervous system
- DH:
-
Dorsal horn
- DIO:
-
Diet-induced obesity
- DRG:
-
Dorsal root ganglion
- FGF21:
-
Fibroblast growth factor 21
- HFD:
-
High-fat diet
- LHA:
-
Lateral hypothalamic area
- LXR:
-
Liver X receptor
- NG:
-
Nodose ganglion
- NGF:
-
Nerve growth factor
- NPY:
-
Neuropeptide Y
- NR:
-
Nuclear receptor
- OT:
-
Oxytocin
- OXTR:
-
Oxytocin receptor
- PAG:
-
Periaqueductal gray
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PNS:
-
Peripheral nervous system
- POA:
-
Preoptic area
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- PRV152:
-
Pseudorabies virus 152
- PSNS:
-
Parasympathetic nervous system
- PVH:
-
Paraventricular hypothalamus
- SNS:
-
Sympathetic nervous system
- SP:
-
Substance P
- TG:
-
Trigeminal ganglion
- TH:
-
Tyrosine hydroxylase
- TRP:
-
Transient receptor potential
- TRPV:
-
Transient receptor potential vanilloid
- UCP1:
-
Uncoupling protein 1
- VMH:
-
Ventromedial hypothalamus
- VNS:
-
Vagus nerve stimulation
- WAT:
-
White adipose tissue
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
The World Health Organization. ISBN: 978 92 4 156485 4 http://www.who.int/nmh/publications/ncd-status-report-2014/en/. Retrieved: March 2016.
Ward ZJ, Long MW, Resch SC, Gortmaker SL, Cradock AL, Giles C, et al. Redrawing the US obesity landscape: bias-corrected estimates of state-specific adult obesity prevalence. PLoS One. 2016;11(3):e0150735.
Townsend KL, Tseng YH. Brown fat fuel utilization and thermogenesis. Trends Endocrinol Metab. 2014;25(4):168–77. Review.
Chondronikola M, Volpi E, Børsheim E, Porter C, Annamalai P, Enerbäck S, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. 2014;63(12):4089–99.
Bamshad M, Song CK, Bartness TJ. CNS origins of the sympathetic nervous system outflow to brown adipose tissue. Am J Physiol. 1999;276(6 Pt 2):R1569–78.
Vaughan CH, Bartness TJ. Anterograde transneuronal viral tract tracing reveals central sensory circuits from brown fat and sensory denervation alters its thermogenic responses. Am J Physiol Regul Integr Comp Physiol. 2012;302(9):R1049–58.
Ryu V, Garretson JT, Liu Y, Vaughan CH, Bartness TJ. Brown adipose tissue has sympathetic-sensory feedback circuits. J Neurosci. 2015;35(5):2181–90.
Ryu V, Bartness TJ. Short and long sympathetic-sensory feedback loops in white fat. Am J Physiol Regul Integr Comp Physiol. 2014;306(12):R886–900.
Bartness TJ, Song CK. Thematic review series: adipocyte biology. Sympathetic and sensory innervation of white adipose tissue. J Lipid Res. 2007;48(8):1655–72.
Youngstrom TG, Bartness TJ. Catecholaminergic innervation of white adipose tissue in Siberian hamsters. Am J Physiol. 1995;268(3 Pt 2):R744–51.
Kreier F, Kap YS, Mettenleiter TC, van Heijningen C, van der Vliet J, Kalsbeek A, et al. Tracing from fat tissue, liver, and pancreas: a neuroanatomical framework for the role of the brain in type 2 diabetes. Endocrinology. 2006;147(3):1140–7.
Fishman RB, Dark J. Sensory innervation of white adipose tissue. Am J Physiol. 1987;253(6 Pt 2):R942–4.
Burneo JG, Faught E, Knowlton R, Morawetz R, Kuzniecky R. Weight loss associated with vagus nerve stimulation. Neurology. 2002;59(3):463–4.
Abubakr A, Wambacq I. Long-term outcome of vagus nerve stimulation therapy in patients with refractory epilepsy. J Clin Neurosci. 2008;15(2):127–9.
Pardo JV, Sheikh SA, Kuskowski MA, Surerus-Johnson C, Hagen MC, Lee JT, et al. Weight loss during chronic, cervical vagus nerve stimulation in depressed patients with obesity: an observation. Int J Obes (Lond). 2007;31(11):1756–9.
Bugajski AJ, Gil K, Ziomber A, Zurowski D, Zaraska W, Thor PJ. Effect of long-term vagal stimulation on food intake and body weight during diet induced obesity in rats. J Physiol Pharmacol. 2007;58 Suppl 1:5–12.
Follesa P, Biggio F, Gorini G, Caria S, Talani G, Dazzi L, et al. Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res. 2007;1179:28–34.
Banni S, Carta G, Murru E, Cordeddu L, Giordano E, Marrosu F, et al. Vagus nerve stimulation reduces body weight and fat mass in rats. PLoS One. 2012;7(9):e44813.
Saindon CS, Blecha F, Musch TI, Morgan DA, Fels RJ, Kenney MJ. Effect of cervical vagotomy on sympathetic nerve responses to peripheral interleukin-1beta. Auton Neurosci. 2001;87(2–3):243–8.
Balbo SL, Grassiolli S, Ribeiro RA, Bonfleur ML, Gravena C, Brito Mdo N, et al. Fat storage is partially dependent on vagal activity and insulin secretion of hypothalamic obese rat. Endocrine. 2007;31(2):142–8.
Mano-Otagiri A, Ohata H, Iwasaki-Sekino A, Nemoto T, Shibasaki T. Ghrelin suppresses noradrenaline release in the brown adipose tissue of rats. J Endocrinol. 2009;201(3):341–9.
Liu C, Bookout AL, Lee S, Sun K, Jia L, Lee C, et al. PPARγ in vagal neurons regulates high-fat diet induced thermogenesis. Cell Metab. 2014;19(4):722–30. Provides a novel mechanism by which vagus neurons help regulate energy balance.
Mansuy-Aubert V, Gautron L, Lee S, Bookout AL, Kusminski C, Sun K, et al. Loss of the liver X receptor LXRα/β in peripheral sensory neurons modifies energy expenditure. Elife. 2015;15:4.
Zheng J. Molecular mechanism of TRP channels. Compr Physiol. 2013;3(1):221–42.
Saito M, Yoneshiro T, Matsushita M. Food ingredients as anti-obesity agents. Trends Endocrinol Metab. 2015;26(11):585–7.
Planells-Cases R, Garcìa-Sanz N, Morenilla-Palao C, Ferrer-Montiel A. Functional aspects and mechanisms of TRPV1 involvement in neurogenic inflammation that leads to thermal hyperalgesia. Pflugers Arch. 2005;451(1):151–9.
Zhang LL, Yan Liu D, Ma LQ, Luo ZD, Cao TB, Zhong J, et al. Activation of transient receptor potential vanilloid type-1 channel prevents adipogenesis and obesity. Circ Res. 2007;100(7):1063–70.
Nathan JD, Patel AA, McVey DC, Thomas JE, Prpic V, Vigna SR, et al. Capsaicin vanilloid receptor-1 mediates substance P release in experimental pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2001;281(5):G1322–8.
Motter AL, Ahern GP. TRPV1-null mice are protected from diet-induced obesity. FEBS Lett. 2008;582(15):2257–62.
Blesch A, Tuszynski MH. GDNF gene delivery to injured adult CNS motor neurons promotes axonal growth, expression of the trophic neuropeptide CGRP, and cellular protection. J Comp Neurol. 2001;436(4):399–410.
Lee E, Jung DY, Kim JH, Patel PR, Hu X, Lee Y, et al. Transient receptor potential vanilloid type-1 channel regulates diet-induced obesity, insulin resistance, and leptin resistance. FASEB J. 2015;29(8):3182–92.
Ohyama K, Nogusa Y, Suzuki K, Shinoda K, Kajimura S, Bannai M. A combination of exercise and capsinoid supplementation additively suppresses diet-induced obesity by increasing energy expenditure in mice. Am J Physiol Endocrinol Metab. 2015;308(4):E315–23.
Saito M. Capsaicin and related food ingredients reducing body fat through the activation of TRP and brown fat thermogenesis. Adv Food Nutr Res. 2015;76:1–28.
Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38(3–4):233–52.
Alawi KM, Aubdool AA, Liang L, Wilde E, Vepa A, Psefteli MP, et al. The sympathetic nervous system is controlled by transient receptor potential vanilloid 1 in the regulation of body temperature. FASEB J. 2015;29(10):4285–98. TRPV1 is involved in thermoregulation.
Luo Z, Ma L, Zhao Z, He H, Yang D, Feng X, et al. TRPV1 activation improves exercise endurance and energy metabolism through PGC-1α upregulation in mice. Cell Res. 2012;22(3):551–64.
Cheung SY, Huang Y, Kwan HY, Chung HY, Yao X. Activation of transient receptor potential vanilloid 3 channel suppresses adipogenesis. Endocrinology. 2015;156(6):2074–86. Activation of TRPV3 channel inhibits adipogenesis in vitro and decreases visceral adiposity in vivo.
Sun W, Uchida K, Suzuki Y, Zhou Y, Kim M, Takayama Y, et al. Lack of TRPV2 impairs thermogenesis in mouse brown adipose tissue. EMBO Rep. 2016;17(3):383–99. Although lack of TRPV2 channel increases brown adipocyte differentiation, it impairs the thermogenic function of the tissue.
Morrison SF, Nakamura K. Central neural pathways for thermoregulation. Front Biosci (Landmark Ed). 2011;16:74–104. Review.
Morrison SF, Madden CJ. Central nervous system regulation of brown adipose tissue. Compr Physiol. 2014;4(4):1677–713. Review.
Sáinz N, Barrenetxe J, Moreno-Aliaga MJ, Martínez JA. Leptin resistance and diet-induced obesity: central and peripheral actions of leptin. Metabolism. 2015;64(1):35–46.
Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev. 2011;91(2):389–411. Review.
Satoh N, Ogawa Y, Katsuura G, Numata Y, Tsuji T, Hayase M, et al. Sympathetic activation of leptin via the ventromedial hypothalamus: leptin-induced increase in catecholamine secretion. Diabetes. 1999;48(9):1787–93.
Enriori PJ, Sinnayah P, Simonds SE, Garcia RC, Cowley MA. J Neurosci. 2011;31:12189.
Pellegrino MJ, McCully BH, Habecker BA. Leptin stimulates sympathetic axon outgrowth. Neurosci Lett. 2014;566:1–5. Demonstrates leptin’s ability to stimulate axonal growth of sympathetic neurons in vitro.
Zeng W, Pirzgalska RM, Pereira MM, Kubasova N, Barateiro A, Seixas E, et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell. 2015;163(1):84–94. Shows leptin-mediated direct synapsing of sympathetic neurons on white adipose tissue in vivo.
Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454(7207):1000–4.
Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, et al. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature. 2013;495(7441):379–83.
Townsend KL, Suzuki R, Huang TL, Jing E, Schulz TJ, Lee K, et al. Bone morphogenetic protein 7 (BMP7) reverses obesity and regulates appetite through a central mTOR pathway. FASEB J. 2012;26(5):2187–96.
Whittle AJ, Carobbio S, Martins L, Slawik M, Hondares E, Vázquez MJ, et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell. 2012;149(4):871–85.
Lipton JM, Glyn JR. Central administration of peptides alters thermoregulation in the rabbit. Peptides. 1980;1(1):15–8.
Oldfield BJ, Allen AM, Davern P, Giles ME, Owens NC. Lateral hypothalamic ‘command neurons’ with axonal projections to regions involved in both feeding and thermogenesis. Eur J Neurosci. 2007;25(8):2404–12.
Kasahara Y, Sato K, Takayanagi Y, Mizukami H, Ozawa K, Hidema S, et al. Oxytocin receptor in the hypothalamus is sufficient to rescue normal thermoregulatory function in male oxytocin receptor knockout mice. Endocrinology. 2013;154(11):4305–15.
Kasahara Y, Takayanagi Y, Kawada T, Itoi K, Nishimori K. Impaired thermoregulatory ability of oxytocin-deficient mice during cold-exposure. Biosci Biotechnol Biochem. 2007;71(12):3122–6.
Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, et al. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab. 2014;20(4):670–7. Provide the mechanism by which pharmacologically delivered FGF21 acts centrally to increase energy expenditure.
Hsuchou H, Pan W, Kastin AJ. The fasting polypeptide FGF21 can enter brain from blood. Peptides. 2007;28(12):2382–6.
Yang C, Jin C, Li X, Wang F, McKeehan WL, Luo Y. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PLoS One. 2012;7(3):e33870.
Sarruf DA, Thaler JP, Morton GJ, German J, Fischer JD, Ogimoto K, et al. Fibroblast growth factor 21 action in the brain increases energy expenditure and insulin sensitivity in obese rats. Diabetes. 2010;59(7):1817–24.
Tan BK, Hallschmid M, Adya R, Kern W, Lehnert H, Randeva HS. Fibroblast growth factor 21 (FGF21) in human cerebrospinal fluid: relationship with plasma FGF21 and body adiposity. Diabetes. 2011;60(11):2758–62.
Bookout AL, de Groot MH, Owen BM, Lee S, Gautron L, Lawrence HL, et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med. 2013;19(9):1147–52.
Kreier F, Fliers E, Voshol PJ, Van Eden CG, Havekes LM, Kalsbeek A, et al. Selective parasympathetic innervation of subcutaneous and intra-abdominal fat—functional implications. J Clin Invest. 2002;110(9):1243–50.
Giordano A, Song CK, Bowers RR, Ehlen JC, Frontini A, Cinti S, et al. White adipose tissue lacks significant vagal innervation and immunohistochemical evidence of parasympathetic innervation. Am J Physiol Regul Integr Comp Physiol. 2006;291(5):R1243–55.
De Matteis R, Ricquier D, Cinti S. TH-, NPY-, SP-, and CGRP-immunoreactive nerves in interscapular brown adipose tissue of adult rats acclimated at different temperatures: an immunohistochemical study. J Neurocytol. 1998;27(12):877–86.
Ruschke K, Ebelt H, Klöting N, Boettger T, Raum K, Blüher M, et al. Defective peripheral nerve development is linked to abnormal architecture and metabolic activity of adipose tissue in Nscl-2 mutant mice. PLoS One. 2009;4(5):e5516.
Wan Y, Xue R, Wang Y, Zhang Q, Huang S, Wu W, et al. The effect of neuropeptide Y on brown-like adipocyte’s differentiation and activation. Peptides. 2015;63:126–33.
Yang K, Guan H, Arany E, Hill DJ, Cao X. Neuropeptide Y is produced in visceral adipose tissue and promotes proliferation of adipocyte precursor cells via the Y1 receptor. FASEB J. 2008;22(7):2452–64.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Magdalena Blaszkiewicz and Kristy L. Townsend declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Obesity Treatment
Rights and permissions
About this article

Cite this article
Blaszkiewicz, M., Townsend, K.L. Adipose Tissue and Energy Expenditure: Central and Peripheral Neural Activation Pathways. Curr Obes Rep 5, 241–250 (2016). https://doi.org/10.1007/s13679-016-0216-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-016-0216-9