
Abstract
Purpose of Review
To summarize the recent literature on the pathophysiology, diagnosis, and treatment of pyoderma gangrenosum.
Recent Findings
A complex interplay between both the innate and adaptive immune systems underlies the pathogenesis of pyoderma gangrenosum (PG). Diagnosis remains a challenge, as there is no gold standard test to confirm the presence of the disease. Efforts to establish diagnostic criteria based on clinical findings have recently been proposed. Definitive management strategies are also lacking; however, current trends in treatment have favored the use of immunosuppressive medications, wound care management, and analgesia.
Summary
PG is a complex disease that continues to pose a challenge. Current research on PG is focused on improving our understanding of the pathophysiology so that we might improve our diagnostic consistency and identify treatment approaches optimized for each individual patient’s specific pathology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pyoderma gangrenosum (PG), a rare autoinflammatory disease considered a prototypic neutrophilic dermatosis, presents as ulcerated lesions most commonly appearing on the lower extremities. It affects approximately three to ten patients per million; however, this might be underestimated due to lack of a diagnostic test and frequent misdiagnosis. The pathogenesis of PG is complex, as it can be idiopathic or present in association with a variety of inflammatory or neoplastic conditions. Occasionally, PG may have extracutaneous manifestations, including sterile neutrophilic infiltration of internal organs, muscle, or bone [1, 2]. Patients with a history of inflammatory bowel disease are especially susceptible to peristomal PG, a subtype that may occur after placement of an ileostomy or colostomy, which comes with its own diagnostic and treatment challenges. Moreover, PG can also occur in the context of autoinflammatory syndromes (PAPA, SAPHO, and PASH).
Classically, patients develop one or more irregularly shaped ulcers with undermined edges that display a characteristic gun-metal gray or violaceous hue. The ulcers typically have differing sizes and depths; frequently, the ulcers extend deeply to expose underlying muscle and tendons. However, non-ulcerative forms have been described including pustular, bullous, and vegetative forms. While systemic corticosteroids or cyclosporine are considered a mainstay of treatment, response to either topical or systemic immunosuppressives can be unpredictable. Recalcitrant cases are not uncommon in clinical practice. Large-scale studies and clinical trials have been limited. Recent studies have focused on gaining a better understanding of the pathogenesis of PG, improving the diagnosis, and finding targeted treatment options. The present review aims to summarize the most recent literature on PG to improve our comprehension of one of the most perplexing diseases in dermatology.
Pathophysiology
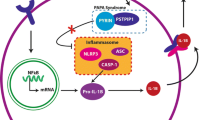
PG is currently considered an autoinflammatory ailment that is most likely secondary to aberrant activation of the innate immune system in patients with a genetic predisposition. It has also been recently proposed that the adaptive immune system might play a role in the pathogenesis of this condition [3•]. It remains unknown how different external triggers (e.g., pathergy, the process through which minor skin trauma leads to ulceration) interact with a patient’s intrinsic genetic factors to cause the varying morphologic presentations of PG. Additionally, it is poorly understood how the interface of these factors then goes on to affect clinical outcomes for patients. We strongly believe that understanding of the pathophysiology of PG is the cornerstone to revealing further diagnostic and therapeutic clues. The current knowledge on the pathogenesis has been summarized in Fig. 1 [3•, 4,5,6,7,8,9,10, 11••, 12,13,14,15, 16•, 17,18,19,20, 21••, 22,23,24,25,26,27,28]. Of note, as indicated in Fig. 1, there is a synergistic interplay between the innate and adaptive immune systems that likely contributes to the pathogenesis of PG. Furthermore, the current understanding of PG places a strong emphasis on the role of IL-1β. Studies have found elevated levels of IL-1β in PG lesions, and several case reports have documented successful treatment with therapies that interfere with IL-1 activity [11••, 16•, 23, 29,30,31,32,33,34]. Additionally, genetic abnormalities documented in syndromic PG also support IL-1 as a potential causative factor. Mutations in the proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1) gene on chromosome 15 have been described in pyogenic arthritis, PG, and acne (PAPA) syndrome and pyogenic arthritis, PG, acne, and hidradenitis suppurativa (PAPASH) syndrome, where decreased inhibition of inflammasomes results in increased IL-1β and IL-18, and subsequently, neutrophilic infiltration [9, 26, 35]. Similarly, increased CCTG microsatellite repeats have been observed in the promoter region of the PSTPIP1 gene in cases of PG, acne, and suppurative hidradenitis (PASH) syndrome [10]. Just as the IL-1 antagonists anakinra and canakinumab have led to improvement in PG, biologics targeting other inflammatory markers (i.e., TNF-α, IL-12/23, IL-6, a4b7 integrin) have also led to ulcer resolution [28, 36,37,38]. It is possible that different etiologies and potentially different phenotypes could be determined based upon responsiveness to blocking certain inflammatory pathways.

Pathogenesis of pyoderma gangrenosum. This schematic diagram addresses the complexity of the pathogenesis of PG. It depicts the influence and interactions of several innate and adaptive genes, as well as a variety of molecules in the immune system that may predispose patients to develop PG lesions. CD, cluster of differentiation; DOCK, dedicator of cytokinesis; G-CSF, granulocyte colony-stimulating factor; GPBAR, G protein-coupled bile acid receptor; IBD, inflammatory bowel disease; IFN, interferon; IL, interleukin; IL-1RN, gene for interleukin 1 receptor antagonist; IL-8RA, gene for interleukin 8 receptor alpha; JAK, Janus kinase; LPIN2, gene for lipin 2; MEFV, gene for Mediterranean fever; MMP, matrix metalloproteinase; MTHFR, methylene tetrahydrofolate reductase; MUC17, gene for mucin 17; NK, natural killer; NLRP, NOD-like receptor protein; NOD, nucleotide-binding oligomerization domain-containing protein; PAPA, pyogenic arthritis, pyoderma gangrenosum, acne; PAPASH, pyogenic arthritis, pyoderma gangrenosum, acne, and suppurative hidradenitis; PASH, pyoderma gangrenosum, acne, and suppurative hidradenitis; PSMB, proteasome subunit beta; PSTPIP, proline-serine-threonine phosphatase-interacting protein; PV, polycythemia vera; RAG, recombination activating gene; RANTES, regulated on activation, normal T cell expressed and secreted; TIMP, tissue inhibitor of metalloproteinase; TLR, Toll-like receptor; TNF, tumor necrosis factor; SCID, severe combined immunodeficiency; TRAF3IP2, TNF receptor-associated factor 3-interacting protein 2; WNK, human gene for WNK lysine-deficient protein kinase
Diagnosis
Misdiagnosing PG is not uncommon in clinical practice. Previous reports have demonstrated a misdiagnosis rate of 10–20% [39]. PG has primarily been a diagnosis of exclusion, and the lack of validated diagnostic criteria [40] has made it difficult to conduct clinical studies on patients with PG. In response to these challenges, several attempts have been made to create formal diagnostic criteria to better distinguish PG from similar ulcerative skin conditions. A recent panel of dermatology experts convened to develop diagnostic criteria utilizing a Delphi approach [41••]. Nine criteria—one major and eight minor—were suggested to assist with diagnosis of PG (Table 1). The presence of the one major criterion and at least four of the eight minor criteria are required for a diagnosis of PG to be made. The selected criteria were validated against 113 case reports of ulcerative PG (n = 65) and PG mimickers (n = 48), which yielded a sensitivity and specificity of 86 and 90%, respectively.
Most recently, a group of European dermatology experts developed a diagnostic tool for PG titled the PARACELSUS score [42••]. The components of this score are based on a combination of criteria that have been previously reported in the literature [40, 43, 44], as well as the findings from a retrospective chart review evaluating the clinical history and images of 60 patients with PG. The astutely named PARACELSUS score consists of ten criteria that are then sub-classified into major, minor, and additional criteria (Table 1). The percentage of patients who presented with each of the key findings was then used to define what constitutes a major, minor, and additional classification; major criteria were defined as being present in > 95% of patients, minor criteria were present in 60–94% of patients, and additional criteria were present in less than 60% of patients. A scoring system was then developed assigning 3 points to major criteria, 2 points to minor criteria, and 1 point to additional criteria. Using the PARACELSUS score, two teams of experts reviewed the clinical history and images of 60 patients with confirmed PG and 50 patients with venous leg ulcers. Compared to the group of patients with venous leg ulcers, all patients with confirmed PG scored ≥ 10 points. Patients with venous leg ulcers all scored ≤ 7 points. To further validate the criteria, alternative definitions of major, minor, and alternative criteria as well as various scoring scales were tested, all of which resulted in comparable findings with PG cases having significantly higher scores than controls.
Both of these newly proposed diagnostic schemes advance the current effort to improve the accuracy and consistency of PG diagnoses. Despite the overlap in several of the proposed criteria (Table 1), the role of skin biopsy was inconsistent between the two diagnostic tools. Importantly, assessing for and excluding other similar appearing ulcerating conditions of the skin are a key step in both approaches. Exclusion of infection by microbiological cultures (via tissue culture or superficial swab), a relevant differential diagnosis to consider, is not explicitly included in the newly proposed criteria for PG diagnosis. Waiting for culture results, which can take upwards of 2 days or more, can postpone initiation of immunosuppression and ultimately lead to untimely, ineffective care. Not infrequently, patients with PG will have endured numerous unsuccessful courses of antibiotics and mechanical debridements and had negative cultures by the time a dermatologist is consulted. As diagnostic molecular microbiology continues to evolve, new techniques to improve the diagnosis of infections in a more expeditious and effective manner might be incorporated in the workup of patients with PG.
Future prospective studies evaluating the utility of the proposed diagnostic criteria should aim to clarify their accuracy and feasibility in the clinical practice setting. Only time will determine how the medical community will apply these criteria in their clinical and research practices; however, we foresee challenges asking non-dermatologists (primary care providers, emergency medicine physicians, surgeons) to use diagnostic criteria/tools, as they do not receive training to recognize the appearance of PG ulcers and they are not as familiar with the diagnosis of atypical ulcers such as PG. A gold standard diagnostic test might fulfill this need but is still lacking.
Treatment
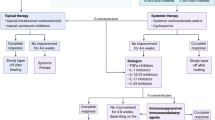
There is no gold standard treatment for PG. However, it is widely accepted that the mainstay treatment requires topical or systemic immunosuppression combined with wound care and pain management (Fig. 2). The authors’ proposed algorithm is also depicted below (Fig. 3).

Treatment approach for patients with PG

PG treatment algorithm. Management of PG frequently requires concurrent use of a variety of immunosuppressive and immunomodulating medications. This figure shows the authors’ preference in regard to combining these medications. Medications in different colored boxes can be safely combined for treatment of PG
Immunosuppression
Systemic corticosteroids and cyclosporine are often the first-line therapy; however, intralesional and topical applications have also been successful in patients with small and/or smoldering ulcerations (based on our experience, small ulcers are < 2 cm). Peristomal PG is also particularly responsive to topical corticosteroids and calcineurin inhibitors, with one review citing complete healing in 62 and 56%, respectively, of patients using these treatments [45]. In 2015, the randomized control trial titled STOP GAP found no significant difference in outcomes among patients who received oral cyclosporine compared to prednisolone. By 6 months, almost 50% of PG ulcers had healed, irrespective of whether patients received oral cyclosporine or prednisolone [46••]. This study also suggested patients with diabetes, osteoporosis, and/or peptic ulceration should avoid systemic corticosteroids, while patients with hypertension or renal insufficiency should not use cyclosporine.
The addition of topical or systemic antimicrobials and anti-neutrophilic agents (e.g., dapsone, colchicine) has been based on physician’s preference. Based on the authors’ experience, our group advocates for the use of adjuvant dapsone in addition to immunosuppression for acute treatment of PG. Dapsone has a dual effect in patients with PG; it has anti-inflammatory actions to inhibit chemotaxis of neutrophils [41••] and provides prophylaxis against Pneumocystis jiroveci (PJP) while the patient is receiving chronic immunosuppression [47].
Recent investigations into alternative treatment options for PG have focused on biologics. With the increasing knowledge of the role of cytokines and other inflammatory molecules in the pathogenesis of PG, the ability to target specific mediators of the disease process has become an increasingly favorable approach. TNF-α inhibitors have been studied in several small samples yielding mixed results [48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. These therapies offer a two-pronged approach to treatment, as about half of patients presenting with PG have a comorbid condition that is also responsive to treatment with these medications. Specifically, infliximab has shown a strong benefit among patients with and without IBD in a randomized clinical trial [36]. Almost half of the patients reported improvement after 2 weeks, and two thirds reported improvement by week 6. The remainder of the patients had no response to treatment. Only a couple of patients developed serious adverse events, which included congestive heart failure (1/29 patients) and MRSA soft tissue infection complicated with sepsis (1/29).
Case reports and small case series have also described high rates of complete healing or significant clinical improvement in patients with syndromic and non-syndromic PG using other TNF inhibitors such as adalimumab [64,65,66,67,68,69,70,71,72,73], certolizumab [74, 75], and etanercept [76,77,78,79,80,81]. Conversely, reports of golimumab therapy have described failure of improvement in severe recalcitrant PG [82] and drug-associated PG in the setting of RA [83]. Another viable alternative to TNF-α inhibitors has been ustekinumab [28, 38, 82, 84], an IL-12/23 antibody, which has been especially effective at higher doses [84]. Tocilizumab, an IL-6 inhibitor, has also led to ulcer resolution in a patient with RA and interstitial lung disease, a contraindication for TNF-α inhibitors [37]. Other beneficial therapies in patients with comorbid IBD include vedolizumab, which interferes with gastrointestinal T cells [38], and visilizumab, an anti-CD3 antibody [85].
There are also some case reports supporting the inhibition of IL-1 as another viable treatment option in patients with PG. Anakinra, an IL-1 receptor antagonist, has been successfully utilized in the treatment of some patients with syndromic and non-syndromic PG [32,33,34, 86,87,88]. On the other hand, there are some cases reporting failure of this approach [89]. Additionally, canakinumab, an IL-1β antagonist, has had success in treating patients with steroid-refractory PG [16•, 29, 30]. In one report, five out of six patients had either a complete or a partial response, and only one patient did not respond to treatment [16•]. Additional case reports have documented treatment failure with canakinumab as well [90].
While there is favorable case-based evidence to support the use of biologics in the treatment of PG, further study with larger sample sizes is warranted before any conclusions can be drawn on their overall effectiveness in patients with syndromic and non-syndromic PG. Notably, their use might be advantageous when patients present with underlying systemic conditions (inflammatory bowel disease, rheumatoid arthritis) or associated skin conditions (hidradenitis suppurativa, psoriasis) that are also responsive to these medications.
More recently, small-molecule drugs have also been considered as potential therapies for patients with PG, including Janus kinase (JAK) inhibitors (e.g., ruxolitinib) [14, 15, 91] and PDE4 inhibitors (apremilast) [92].
Based on the authors’ experience, high doses of intravenous immunoglobulin (IVIG), with 2 g/kg administered over 2 to 3 consecutive days once a month for 6 months, have been another useful alternative. This is particularly helpful in recalcitrant cases and in cases with repetitive superinfection of PG ulcers. It has been proposed that the effect of IVIG is likely due to its anti-inflammatory activity, which involves decreasing the half-life of IgG antibodies by binding the neonatal Fc receptor (FcRn), inhibition of Fc receptor activation, and prevention of tissue destruction by complement [93, 94]. Additionally, if a patient has contraindications to the aforementioned medications due to comorbidities and immunosuppression, IVIG seems to have a relatively safe profile [94].
Wound Care Management
Wound care is another key component of PG management that should be used in conjunction with immunosuppressive therapy. Creating an appropriate environment that will foster re-vascularization and re-epithelialization is crucial for the healing of PG ulcers. Dressings are especially important in preventing potential superinfections. Several types of dressings have been reported in the treatment of PG ulcers including hydrogels and films, non-adherent povidone-iodine (Inadine™; Systagenix) [95]; alginate [95]; acellular bovine collagen-glycosamine complex/silicone (Integra™ Matrix Wound Dressing) [96]; flexible polyester mesh impregnated with hydrocolloid and petroleum jelly particles (UrgoTul®; Urgo Medical) [97]; sodium carboxymethylcellulose (NaCMC) containing 1.2% silver (Aquacel® Silver) [97]; antimicrobial foam (Mepilex® Ag; Mölnlycke) [97]; sulfamylon [98]; 45% oxidized regenerated cellulose and 55% collagen composite (PROMOGRAN™ Matrix; Acelity) [99]; nanocrystalline silver alginate (Acticoat Absorbent®; Smith & Nephew) [99]; and lyophilized type I bovine collagen matrix (SkinTemp®; Biocore Inc.) [100]. The dressing selected depends on the ulcer’s characteristics, which includes the amount of drainage, the presence of fibrin/slough, and the presence of non-viable tissue. Sharp debridement is not usually recommended in PG ulcers; however, if the presence of non-viable tissue is impairing healing and the patient is currently receiving immunosuppression, debridement becomes another alternative to improve the ulcer environment. Moreover, there is some evidence for the use of negative-pressure wound therapy in conjunction with systemic immunosuppression [101, 102], which might represent another alternative to promote healing. Some cases have reported successful use of adjuvant hyperbaric oxygen [103,104,105,106]. Biologic dressings might represent an alternative to accelerate the healing process [107, 108], but this is still an unexplored area of research. Finally, wireless microcurrent stimulation has also been reported as beneficial in a small number of PG patients [109].
Surgery has traditionally been considered counterproductive to PG treatment, as the trauma introduced to the wound may lead to a pathergy reaction facilitating further ulceration. However, based on a systematic review, there may be a role for surgical treatment using negative-pressure wound therapy (NPWT) followed by split-thickness skin graft (STSG) alongside antibiotic prophylaxis and other adjuvant immunosuppressive/immunomodulatory treatment as needed. This approach led to increased healing rates in the majority of patients studied [110]. Despite the favorable outcomes observed, it must be noted that STSG does confer a risk of relapse or pathergy; however, this risk could be minimized if the patient is receiving concurrent systemic immunosuppression. A proposed lower risk alternative to STSG is epidermal grafting in which grafts are harvested via suction blisters created by heat and negative pressure. One study reports complete healing in three out of five patients and reduction in ulcer size in two others who underwent this procedure; no pathergy was observed [111]. Unfortunately, amputation has also been performed and reported in some patients with severe recalcitrant PG [95, 106, 112].
Pain Management
Pain control is often necessary in conjunction with wound care and typically consists of systemic anti-inflammatories or opioids. Topical morphine has also been used with some success in patients with PG and other chronic ulcers [113, 114]. In addition to opioids, combination therapy with neuropathic medications (gabapentin or pregabalin) or antidepressants has been suggested to address neuropathic pain, which can develop secondary to nerve damage from ulceration [115]. Interestingly, newer evidence suggests opioids are associated with decreased healing rates in venous ulcers; therefore, alternative pain management strategies are being considered [116]. A recent report documents clinically significant pain reduction using topical medical cannabis in three patients whose pain was uncontrolled with opioids and acetaminophen [117].
Additional general therapeutic measurements include minimizing edema with compression garments, smoking cessation, glycemic control in diabetics, and optimizing nutritional status [35].
Conclusion
Pyoderma gangrenosum is a complex autoinflammatory ulcerative skin condition. It is a rare pathology for which the etiology has yet to be fully understood. A number of inflammatory mediators have been identified as playing a role in its pathogenesis, and a growing body of evidence suggests both innate and adaptive immune cells malfunction to initiate the disease process. Indeed, the complex interaction between genetic predisposition and immune dysfunction makes it possible that no single causative pathway exists, but instead, under the right conditions, a number of stimuli may trigger the cascade of inflammation. Furthermore, the incomplete understanding of the pathophysiology has also posed challenges in diagnosis and treatment. No biomarkers exist to detect PG; therefore, diagnosis has traditionally been made on the basis of exclusion. Fortunately, promising diagnostic criteria have recently been proposed to attempt to increase accuracy and consistency in diagnosis among medical providers. Recent attention has also been directed toward developing an outcome instrument to assess severity and response to treatment. Systemic corticosteroids and cyclosporine have traditionally been the first-line therapies; however, biologics that target the many inflammatory molecules found in PG lesions are now being considered. Moreover, IVIG has been successfully reported in the majority of recalcitrant cases. Collaborations through larger clinical trials are needed before a gold standard treatment protocol is set. In the near future, PG patients will require a precision medicine approach; utilizing genetic inflammatory markers for diagnosis of the different phenotypes to select the appropriate treatment alternatives and to predict outcomes.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Scherlinger M, Guillet S, Doutre MS, Beylot-Barry M, Pham-Ledard A. Pyoderma gangrenosum with extensive pulmonary involvement. J Eur Acad Dermatol Venereol. 2017;31(4):e214–e6. https://doi.org/10.1111/jdv.13976.
Vadillo M, Jucgla A, Podzamczer D, Rufi G, Domingo A. Pyoderma gangrenosum with liver, spleen and bone involvement in a patient with chronic myelomonocytic leukaemia. Br J Dermatol. 1999;141(3):541–3.
• Wang EA, Steel A, Luxardi G, Mitra A, Patel F, Cheng MY, et al. Classic ulcerative pyoderma gangrenosum is a T cell-mediated disease targeting follicular adnexal structures: a hypothesis based on molecular and clinicopathologic studies. Front Immunol. 2017;8:1980. https://doi.org/10.3389/fimmu.2017.01980. This study highlights the potential role of the T cell response directed against pilosebaceous units in patients with PG.
DeFilippis EM, Feldman SR, Huang WW. The genetics of pyoderma gangrenosum and implications for treatment: a systematic review. Br J Dermatol. 2015;172(6):1487–97. https://doi.org/10.1111/bjd.13493.
Antiga E, Maglie R, Volpi W, Bianchi B, Berti E, Marzano AV, et al. T helper type 1-related molecules as well as interleukin-15 are hyperexpressed in the skin lesions of patients with pyoderma gangrenosum. Clin Exp Immunol. 2017;189(3):383–91. https://doi.org/10.1111/cei.12989.
Quaglino P, Fava P, Caproni M, Antiga E, De Simone C, Papini M, et al. Phenotypical characterization of circulating cell subsets in pyoderma gangrenosum patients: the experience of the Italian immuno-pathology group. J Eur Acad Dermatol Venereol. 2016;30(4):655–8. https://doi.org/10.1111/jdv.13100.
Marzano AV, Cugno M, Trevisan V, Fanoni D, Venegoni L, Berti E, et al. Role of inflammatory cells, cytokines and matrix metalloproteinases in neutrophil-mediated skin diseases. Clin Exp Immunol. 2010;162(1):100–7. https://doi.org/10.1111/j.1365-2249.2010.04201.x.
Nesterovitch AB, Hoffman MD, Simon M, Petukhov PA, Tharp MD, Glant TT. Mutations in the PSTPIP1 gene and aberrant splicing variants in patients with pyoderma gangrenosum. Clin Exp Dermatol. 2011;36(8):889–95. https://doi.org/10.1111/j.1365-2230.2011.04137.x.
Demidowich AP, Freeman AF, Kuhns DB, Aksentijevich I, Gallin JI, Turner ML, et al. Brief report: genotype, phenotype, and clinical course in five patients with PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum, and acne). Arthritis Rheum. 2012;64(6):2022–7. https://doi.org/10.1002/art.34332.
Braun-Falco M, Kovnerystyy O, Lohse P, Ruzicka T. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66(3):409–15. https://doi.org/10.1016/j.jaad.2010.12.025.
•• Marzano AV, Damiani G, Ceccherini I, Berti E, Gattorno M, Cugno M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol. 2017;176(6):1588–98. https://doi.org/10.1111/bjd.15226. This study supports the role of the innate immune system and its mutations in syndromic and non-syndromic forms of pyoderma gangrenosum.
Caproni M, Antiga E, Volpi W, Verdelli A, Venegoni L, Quaglino P, et al. The Treg/Th17 cell ratio is reduced in the skin lesions of patients with pyoderma gangrenosum. Br J Dermatol. 2015;173(1):275–8. https://doi.org/10.1111/bjd.13670.
Palanivel JA, Macbeth AE, Levell NJ. Pyoderma gangrenosum in association with Janus kinase 2 (JAK2V617F) mutation. Clin Exp Dermatol. 2013;38(1):44–6. https://doi.org/10.1111/j.1365-2230.2012.04375.x.
Nasifoglu S, Heinrich B, Welzel J. Successful therapy of pyoderma gangrenosum with a Janus kinase 2 inhibitor. Br J Dermatol. 2018. https://doi.org/10.1111/bjd.16468.
Shanmugam VK, McNish S, Shara N, Hubley KJ, Kallakury B, Dunning DM, et al. Chronic leg ulceration associated with polycythemia vera responding to ruxolitinib (Jakafi((R))). J Foot Ankle Surg. 2013;52(6):781–5. https://doi.org/10.1053/j.jfas.2013.07.003.
• Kolios AG, Maul JT, Meier B, Kerl K, Traidl-Hoffmann C, Hertl M, et al. Canakinumab in adults with steroid-refractory pyoderma gangrenosum. Br J Dermatol. 2015;173(5):1216–23. https://doi.org/10.1111/bjd.14037. This study confirms the pathogenic role of IL-1β in pyoderma and proposes the use of canakinumab as a viable alternative to treat recalcitrant cases.
Kawakami T, Yamazaki M, Soma Y. Reduction of interleukin-6, interleukin-8, and anti-phosphatidylserine-prothrombin complex antibody by granulocyte and monocyte adsorption apheresis in a patient with pyoderma gangrenosum and ulcerative colitis. Am J Gastroenterol. 2009;104(9):2363–4. https://doi.org/10.1038/ajg.2009.271.
Kozono K, Nakahara T, Kikuchi S, Itoh E, Kido-Nakahara M, Furue M. Pyoderma gangrenosum with increased levels of serum cytokines. J Dermatol. 2015;42(12):1186–8. https://doi.org/10.1111/1346-8138.12970.
Patiroglu T, Akar HH, Gilmour K, Ozdemir MA, Bibi S, Henriquez F, et al. Atypical severe combined immunodeficiency caused by a novel homozygous mutation in Rag1 gene in a girl who presented with pyoderma gangrenosum: a case report and literature review. J Clin Immunol. 2014;34(7):792–5. https://doi.org/10.1007/s10875-014-0077-5.
Wu BC, Patel ED, Ortega-Loayza AG. Drug-induced pyoderma gangrenosum: a model to understand the pathogenesis of pyoderma gangrenosum. Br J Dermatol. 2017;177(1):72–83. https://doi.org/10.1111/bjd.15193.
•• Ortega-Loayza AG, Nugent WH, Lucero OM, Washington SL, Nunley JR, Walsh SW. Dysregulation of inflammatory gene expression in lesional and nonlesional skin of patients with pyoderma gangrenosum. Br J Dermatol. 2018;178(1):e35–e6. https://doi.org/10.1111/bjd.15837. This study supports the role of pattern recognition receptors in the pathogenesis of pyoderma gangrenosum.
Bister V, Makitalo L, Jeskanen L, Saarialho-Kere U. Expression of MMP-9, MMP-10 and TNF-alpha and lack of epithelial MMP-1 and MMP-26 characterize pyoderma gangrenosum. J Cutan Pathol. 2007;34(12):889–98. https://doi.org/10.1111/j.1600-0560.2007.00744.x.
Marzano AV, Fanoni D, Antiga E, Quaglino P, Caproni M, Crosti C, et al. Expression of cytokines, chemokines and other effector molecules in two prototypic autoinflammatory skin diseases, pyoderma gangrenosum and Sweet's syndrome. Clin Exp Immunol. 2014;178(1):48–56. https://doi.org/10.1111/cei.12394.
Oka M, Berking C, Nesbit M, Satyamoorthy K, Schaider H, Murphy G, et al. Interleukin-8 overexpression is present in pyoderma gangrenosum ulcers and leads to ulcer formation in human skin xenografts. Lab Invest. 2000;80(4):595–604.
Marzano AV, Ortega-Loayza AG, Ceccherini I, Cugno M. LPIN2 gene mutation in a patient with overlapping neutrophilic disease (pyoderma gangrenosum and aseptic abscess syndrome). JAAD Case Rep. 2018;4(2):120–2. https://doi.org/10.1016/j.jdcr.2017.08.020.
Marzano AV, Trevisan V, Gattorno M, Ceccherini I, De Simone C, Crosti C. Pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa (PAPASH): a new autoinflammatory syndrome associated with a novel mutation of the PSTPIP1 gene. JAMA Dermatol. 2013;149(6):762–4. https://doi.org/10.1001/jamadermatol.2013.2907.
Kanameishi S, Nakamizo S, Endo Y, Fujisawa A, Dainichi T, Tanaka T, et al. High level of serum human interleukin-18 in a patient with pyogenic arthritis, pyoderma gangrenosum and acne syndrome. J Eur Acad Dermatol Venereol. 2017;31(2):e115–e6. https://doi.org/10.1111/jdv.13856.
Guenova E, Teske A, Fehrenbacher B, Hoerber S, Adamczyk A, Schaller M, et al. Interleukin 23 expression in pyoderma gangrenosum and targeted therapy with ustekinumab. Arch Dermatol. 2011;147(10):1203–5. https://doi.org/10.1001/archdermatol.2011.168.
Geusau A, Mothes-Luksch N, Nahavandi H, Pickl WF, Wise CA, Pourpak Z, et al. Identification of a homozygous PSTPIP1 mutation in a patient with a PAPA-like syndrome responding to canakinumab treatment. JAMA Dermatol. 2013;149(2):209–15. https://doi.org/10.1001/2013.jamadermatol.717.
Jaeger T, Andres C, Grosber M, Zirbs M, Hein R, Ring J, et al. Pyoderma gangrenosum and concomitant hidradenitis suppurativa--rapid response to canakinumab (anti-IL-1beta). Eur J Dermatol. 2013;23(3):408–10. https://doi.org/10.1684/ejd.2013.2018.
Galimberti RL, Vacas AS, Bollea Garlatti ML, Torre AC. The role of interleukin-1beta in pyoderma gangrenosum. JAAD Case Rep. 2016;2(5):366–8. https://doi.org/10.1016/j.jdcr.2016.07.007.
Beynon C, Chin MF, Hunasehally P, Bhagwandas K, Bevan M, Taylor M, et al. Successful treatment of autoimmune disease-associated pyoderma gangrenosum with the IL-1 receptor antagonist anakinra: a case series of 3 patients. J Clin Rheumatol. 2017;23(3):181–3. https://doi.org/10.1097/rhu.0000000000000511.
Jennings L, Molloy O, Quinlan C, Kelly G, O'Kane M. Treatment of pyoderma gangrenosum, acne, suppurative hidradenitis (PASH) with weight-based anakinra dosing in a hepatitis B carrier. Int J Dermatol. 2017;56(6):e128–e9. https://doi.org/10.1111/ijd.13528.
Mercuri SR, Paolino G, De Flammineis E, Didona D, Brianti P. Successful treatment of pyoderma gangrenosum with anakinra in a patient with Wiskott-Aldrich syndrome. Dermatol Ther. 2018;31(2):e12582. https://doi.org/10.1111/dth.12582.
Alavi A, French LE, Davis MD, Brassard A, Kirsner RS. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18(3):355–72. https://doi.org/10.1007/s40257-017-0251-7.
Brooklyn TN, Dunnill MG, Shetty A, Bowden JJ, Williams JD, Griffiths CE, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55(4):505–9. https://doi.org/10.1136/gut.2005.074815.
Lee WS, Choi YJ, Yoo WH. Use of tocilizumab in a patient with pyoderma gangrenosum and rheumatoid arthritis. J Eur Acad Dermatol Venereol. 2017;31(2):e75–e7. https://doi.org/10.1111/jdv.13736.
Fleisher M, Marsal J, Lee SD, Frado LE, Parian A, Korelitz BI, et al. Effects of vedolizumab therapy on extraintestinal manifestations in inflammatory bowel disease. Dig Dis Sci. 2018;63(4):825–33. https://doi.org/10.1007/s10620-018-4971-1.
Weenig RH, Davis MD, Dahl PR, Su WP. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347(18):1412–8. https://doi.org/10.1056/NEJMoa013383.
Su WP, Davis MD, Weenig RH, Powell FC, Perry HO. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43(11):790–800. https://doi.org/10.1111/j.1365-4632.2004.02128.x.
•• Maverakis E, Ma C, Shinkai K, Fiorentino D, Callen JP, Wollina U, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154(4):461–6. https://doi.org/10.1001/jamadermatol.2017.5980. This study describes the use of the Delphi technique to define cases of pyoderma gangrenosum.
•• Jockenhofer F, Wollina U, Salva KA, Benson S, Dissemond J. The PARACELSUS score: a novel diagnostic tool for pyoderma gangrenosum. Br J Dermatol. 2018. https://doi.org/10.1111/bjd.16401. This study proposes a new diagnostic clinical instrument to improve the diagnosis of pyoderma gangrenosum.
Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34(3):395–409. quiz 10-2
von den Driesch P. Pyoderma gangrenosum: a report of 44 cases with follow-up. Br J Dermatol. 1997;137(6):1000–5.
Afifi L, Sanchez IM, Wallace MM, Braswell SF, Ortega-Loayza AG, Shinkai K. Diagnosis and management of peristomal pyoderma gangrenosum: a systematic review. J Am Acad Dermatol. 2018;78(6):1195–204.e1. https://doi.org/10.1016/j.jaad.2017.12.049.
•• Ormerod AD, Thomas KS, Craig FE, Mitchell E, Greenlaw N, Norrie J, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958. https://doi.org/10.1136/bmj.h2958. This randomized control trial compares the effectiveness of systemic methylprednisolone and cyclosporine to treat patients with pyoderma gangrenosum, concluding both medications should be considered comparable first-line options.
Rodriguez M, Fishman JA. Prevention of infection due to Pneumocystis spp. in human immunodeficiency virus-negative immunocompromised patients. Clin Microbiol Rev. 2004;17(4):770–82, table of contents. https://doi.org/10.1128/cmr.17.4.770-782.2004.
Andrisani G, Guidi L, Papa A, Potenza AE, Cervelli D, Armuzzi A. A case of pyoderma gangrenosum with ulcerative colitis treated with combined approach: infliximab and surgery. J Crohn’s Colitis. 2013;7(5):421–6. https://doi.org/10.1016/j.crohns.2012.07.021.
Bobbitt SA, Klaus EM, Stringer E, Chowdhury D, Finlayson L. Treatment of refractory pyoderma gangrenosum with infliximab in a 17-month-old boy. Dermatol Online J. 2016;22(4).
Chatzinasiou F, Polymeros D, Panagiotou M, Theodoropoulos K, Rigopoulos D. Generalized pyoderma gangrenosum associated with ulcerative colitis: successful treatment with infliximab and azathioprine. Acta Dermatovenerol Croat. 2016;24(1):83–5.
Crouse L, McShane D, Morrell DS, Wu EY. Pyoderma gangrenosum in an infant: a case report and review of the literature. Pediatr Dermatol. 2018. https://doi.org/10.1111/pde.13471.
Groleau PF, Grossberg AL, Gaspari AA. Hidradenitis suppurativa and concomitant pyoderma gangrenosum treated with infliximab. Cutis. 2015;95(6):337–42.
Kokavec J, Rajak S, Huilgol S, Selva D. Pyoderma gangrenosum of the eyelid. Can J Ophthalmol. 2016;51(2):e58–60. https://doi.org/10.1016/j.jcjo.2015.12.004.
Mooij JE, van Rappard DC, Mekkes JR. Six patients with pyoderma gangrenosum successfully treated with infliximab. Int J Dermatol. 2013;52(11):1418–20. https://doi.org/10.1111/j.1365-4632.2011.05201.x.
Patier de la Pena JL, Moreno-Cobo MA, Sanchez-Conde M, Echaniz Quintana AM. Behcet disease and refractory pyoderma gangrenosum with response to infliximab. Rev Clin Esp. 2015;215(1):66–7. https://doi.org/10.1016/j.rce.2014.09.006.
Staub J, Pfannschmidt N, Strohal R, Braun-Falco M, Lohse P, Goerdt S, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29(11):2243–7. https://doi.org/10.1111/jdv.12765.
Teich N, Klugmann T. Rapid improvement of refractory pyoderma gangrenosum with infliximab gel in a patient with ulcerative colitis. J Crohn's Colitis. 2014;8(1):85–6. https://doi.org/10.1016/j.crohns.2013.06.003.
Vahlquist A, Hakansson LD, Ronnblom L, Karawajczyk M, Fasth A, van Gijn ME, et al. Recurrent pyoderma gangrenosum and cystic acne associated with leucocyte adhesion deficiency due to novel mutations in ITGB2: successful treatment with infliximab and adalimumab. Acta Derm Venereol. 2015;95(3):349–51. https://doi.org/10.2340/00015555-1929.
Vestita M, Guida S, Mazzoccoli S, Loconsole F, Foti C. Late paradoxical development of pyoderma gangrenosum in a psoriasis patient treated with infliximab. Eur J Dermatol. 2015;25(3):272–3. https://doi.org/10.1684/ejd.2015.2526.
Zampeli VA, Lippert U, Nikolakis G, Makrantonaki E, Tzellos TG, Krause U, et al. Disseminated refractory pyoderma gangraenosum during an ulcerative colitis flare. Treatment with infliximab. Journal of Dermatological Case Reports. 2015;9(3):62–6. https://doi.org/10.3315/jdcr.2015.1206.
Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor alpha therapy. J Clin Rheumatol. 2012;18(8):413–5. https://doi.org/10.1097/RHU.0b013e318278b84c.
Carrasco Cubero C, Ruiz Tudela MM, Salaberri Maestrojuan JJ, Perez Venegas JJ. Pyoderma gangrenosum associated with inflammatory bowel disease. Report of two cases with good response to infliximab. Reumatol Clin. 2012;8(2):90–2. https://doi.org/10.1016/j.reuma.2011.07.003.
Ueda M, Katoh M, Tanizaki H, Tanioka M, Matsumura Y, Miyachi Y. Refractory pyoderma gangrenosum associated with ulcerative colitis successfully treated with infliximab. Dermatol Online J. 2012;18(1):12.
Carinanos I, Acosta MB, Domenech E. Adalimumab for pyoderma gangrenosum associated with inflammatory bowel disease. Inflamm Bowel Dis. 2011;17(12):E153–4. https://doi.org/10.1002/ibd.21723.
Fonder MA, Cummins DL, Ehst BD, Anhalt GJ, Meyerle JH. Adalimumab therapy for recalcitrant pyoderma gangrenosum. J Burns Wounds. 2006;5:e8.
Heffernan MP, Anadkat MJ, Smith DI. Adalimumab treatment for pyoderma gangrenosum. Arch Dermatol. 2007;143(3):306–8. https://doi.org/10.1001/archderm.143.3.306.
Hubbard VG, Friedmann AC, Goldsmith P. Systemic pyoderma gangrenosum responding to infliximab and adalimumab. Br J Dermatol. 2005;152(5):1059–61. https://doi.org/10.1111/j.1365-2133.2005.06467.x.
Lee H, Park SH, Kim SK, Choe JY, Park JS. Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome (PAPA syndrome) with E250K mutation in CD2BP1 gene treated with the tumor necrosis factor inhibitor adalimumab. Clin Exp Rheumatol. 2012;30(3):452.
Murphy B, Morrison G, Podmore P. Successful use of adalimumab to treat pyoderma gangrenosum, acne and suppurative hidradenitis (PASH syndrome) following colectomy in ulcerative colitis. Int J Color Dis. 2015;30(8):1139–40. https://doi.org/10.1007/s00384-014-2110-9.
Pomerantz RG, Husni ME, Mody E, Qureshi AA. Adalimumab for treatment of pyoderma gangrenosum. Br J Dermatol. 2007;157(6):1274–5. https://doi.org/10.1111/j.1365-2133.2007.08212.x.
Reddick CL, Singh MN, Chalmers RJ. Successful treatment of superficial pyoderma gangrenosum associated with hidradenitis suppurativa with adalimumab. Dermatol Online J. 2010;16(8):15.
Sagami S, Ueno Y, Tanaka S, Nagai K, Hayashi R, Chayama K. Successful use of adalimumab for treating pyoderma gangrenosum with ulcerative colitis under corticosteroid-tapering conditions. Intern Med. 2015;54(17):2167–72. https://doi.org/10.2169/internalmedicine.54.4853.
Zold E, Nagy A, Devenyi K, Zeher M, Barta Z. Successful use of adalimumab for treating fistulizing Crohn’s disease with pyoderma gangrenosum: two birds with one stone. World J Gastroenterol. 2009;15(18):2293–5.
Hurabielle C, Schneider P, Baudry C, Bagot M, Allez M, Viguier M. Certolizumab pegol—a new therapeutic option for refractory disseminated pyoderma gangrenosum associated with Crohn’s disease. J Dermatolog Treat. 2016;27(1):67–9. https://doi.org/10.3109/09546634.2015.1034075.
Cinotti E, Labeille B, Perrot JL, Pallot-Prades B, Cambazard F. Certolizumab for the treatment of refractory disseminated pyoderma gangrenosum associated with rheumatoid arthritis. Clin Exp Dermatol. 2014;39(6):750–1. https://doi.org/10.1111/ced.12393.
Charles CA, Leon A, Banta MR, Kirsner RS. Etanercept for the treatment of refractory pyoderma gangrenosum: a brief series. Int J Dermatol. 2007;46(10):1095–9. https://doi.org/10.1111/j.1365-4632.2007.03286.x.
Disla E, Quayum B, Cuppari GG, Pancorbo R. Successful use of etanercept in a patient with pyoderma gangrenosum complicating rheumatoid arthritis. J Clin Rheumatol. 2004;10(1):50–2. https://doi.org/10.1097/01.rhu.0000111315.94725.9c.
Goldenberg G, Jorizzo JL. Use of etanercept in treatment of pyoderma gangrenosum in a patient with autoimmune hepatitis. J Dermatolog Treat. 2005;16(5–6):347–9. https://doi.org/10.1080/09546630500424722.
McGowan JW, Johnson CA, Lynn A. Treatment of pyoderma gangrenosum with etanercept. J Drugs Dermatol. 2004;3(4):441–4.
Pastor N, Betlloch I, Pascual JC, Blanes M, Banuls J, Silvestre JF. Pyoderma gangrenosum treated with anti-TNF alpha therapy (etanercept). Clin Exp Dermatol. 2006;31(1):152–3. https://doi.org/10.1111/j.1365-2230.2005.01972.x.
Roy DB, Conte ET, Cohen DJ. The treatment of pyoderma gangrenosum using etanercept. J Am Acad Dermatol. 2006;54(3 Suppl 2):S128–34. https://doi.org/10.1016/j.jaad.2005.10.058.
Goldminz AM, Botto NC, Gottlieb AB. Severely recalcitrant pyoderma gangrenosum successfully treated with ustekinumab. J Am Acad Dermatol. 2012;67(5):e237–8. https://doi.org/10.1016/j.jaad.2012.04.045.
Skalkou A, Manoli SM, Sachinidis A, Ntouros V, Petidis K, Pagkopoulou E, et al. Pyoderma gangrenosum and pyogenic arthritis presenting as severe sepsis in a rheumatoid arthritis patient treated with golimumab. Rheumatol Int. 2018;38(1):161–7. https://doi.org/10.1007/s00296-017-3861-8.
Greb JE, Gottlieb AB, Goldminz AM. High-dose ustekinumab for the treatment of severe, recalcitrant pyoderma gangrenosum. Dermatol Ther. 2016;29(6):482–3. https://doi.org/10.1111/dth.12387.
Lorincz M, Kleszky M, Szaloki T Jr, Szaloki T. Pyoderma gangrenosum treated successfully with visilizumab in patients with ulcerative colitis. Orv Hetil. 2010;151(4):144–7. https://doi.org/10.1556/oh.2010.28786.
Dierselhuis MP, Frenkel J, Wulffraat NM, Boelens JJ. Anakinra for flares of pyogenic arthritis in PAPA syndrome. Rheumatology (Oxford). 2005;44(3):406–8. https://doi.org/10.1093/rheumatology/keh479.
Brenner M, Ruzicka T, Plewig G, Thomas P, Herzer P. Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br J Dermatol. 2009;161(5):1199–201. https://doi.org/10.1111/j.1365-2133.2009.09404.x.
Acquitter M, Plantin P, Kupfer I, Auvinet H, Marhadour T. Anakinra improves pyoderma gangrenosum in psoriatic arthritis: a case report. Ann Intern Med. 2015;163(1):70–1. https://doi.org/10.7326/l15-5107.
Lin Z, Hegarty JP, Lin T, Ostrov B, Wang Y, Yu W, et al. Failure of anakinra treatment of pyoderma gangrenosum in an IBD patient and relevance to the PSTPIP1 gene. Inflamm Bowel Dis. 2011;17(6):E41–2. https://doi.org/10.1002/ibd.21684.
Sun NZ, Ro T, Jolly P, Sayed CJ. Non-response to interleukin-1 antagonist canakinumab in two patients with refractory pyoderma gangrenosum and hidradenitis suppurativa. J Clin Aesthet Dermatol. 2017;10(9):36–8.
Alves de Medeiros AK, Speeckaert R, Desmet E, Van Gele M, De Schepper S, Lambert J. JAK3 as an emerging target for topical treatment of inflammatory skin diseases. PLoS One. 2016;11(10):e0164080. https://doi.org/10.1371/journal.pone.0164080.
Laird ME, Tong LX, Lo Sicco KI, Kim RH, Meehan SA, Franks AG Jr. Novel use of apremilast for adjunctive treatment of recalcitrant pyoderma gangrenosum. JAAD Case Rep. 2017;3(3):228–9. https://doi.org/10.1016/j.jdcr.2017.02.019.
Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol. 2008;26:513–33. https://doi.org/10.1146/annurev.immunol.26.021607.090232.
Song H, Lahood N, Mostaghimi A. Intravenous immunoglobulin as adjunct therapy for refractory pyoderma gangrenosum: systematic review of cases and case series. Br J Dermatol. 2018;178(2):363–8. https://doi.org/10.1111/bjd.15850.
Ratnagobal S, Sinha S. Pyoderma gangrenosum: guideline for wound practitioners. J Wound Care. 2013;22(2):68–73. https://doi.org/10.12968/jowc.2013.22.2.68.
Leitsch S, Koban KC, Pototschnig A, Titze V, Giunta RE. Multimodal therapy of ulcers on the dorsum of the hand with exposed tendons caused by pyoderma gangrenosum. Wounds. 2016;28(3):E10–3.
Tay DZ, Tan KW, Tay YK. Pyoderma gangrenosum: a commonly overlooked ulcerative condition. J Family Med Prim Care. 2014;3(4):374–8. https://doi.org/10.4103/2249-4863.148113.
Goshtasby PH, Chami RG, Johnson RM. A novel approach to the management of pyoderma gangrenosum complicating reduction mammaplasty. Aesthet Surg J. 2010;30(2):186–93. https://doi.org/10.1177/1090820x10366011.
Coelho S, Amarelo M, Ryan S, Reddy M, Sibbald RG. Rheumatoid arthritis-associated inflammatory leg ulcers: a new treatment for recalcitrant wounds. Int Wound J. 2004;1(1):81–4. https://doi.org/10.1111/j.1742-481x.2004.0002.x.
Farris DR, Schutzer PJ, Don PC, Silverberg NB, Weinberg JM. Resolution of pyoderma gangrenosum after therapy with lyophilized bovine collagen matrix. Dermatology. 2003;206(3):284–5. https://doi.org/10.1159/000068887.
Tanini S, Calugi G, Russo GL. Combination of negative pressure wound therapy and systemic steroid therapy in postsurgical pyoderma gangrenosum after reduction mammoplasty; a case of proven efficacy and safety. Dermatol Reports. 2017;9(2):7209. https://doi.org/10.4081/dr.2017.7209.
Yamaguchi Y, Yanagi T, Sato K, Yoshimoto N, Hirata Y, Ujiie I, et al. Portable negative-pressure wound therapy for pyoderma gangrenosum: report of two cases. J Dermatol. 2018;45(4):483–6. https://doi.org/10.1111/1346-8138.14180.
Seo HI, Lee HJ, Han KH. Hyperbaric oxygen therapy for pyoderma gangrenosum associated with ulcerative colitis. Intest Res. 2018;16(1):155–7. https://doi.org/10.5217/ir.2018.16.1.155.
Chiang IH, Liao YS, Dai NT, Chiao HY, Chou CY, Chen SG, et al. Hyperbaric oxygen therapy for the adjunctive treatment of pyoderma gangrenosum: a case report. Ostomy Wound Manage. 2016;62(5):32–6.
Tutrone WD, Green K, Weinberg JM, Caglar S, Clarke D. Pyoderma gangrenosum: dermatologic application of hyperbaric oxygen therapy. J Drugs Dermatol. 2007;6(12):1214–9.
Cabalag MS, Wasiak J, Lim SW, Raiola FB. Inpatient management of pyoderma gangrenosum: treatments, outcomes, and clinical implications. Ann Plast Surg. 2015;74(3):354–60. https://doi.org/10.1097/SAP.0b013e31829565f3.
Anselmo DS, McGuire JB, Love E, Vlahovic T. Application of viable cryopreserved human placental membrane grafts in the treatment of wounds of diverse etiologies: a case series. Wounds. 2018;30(3):57–61.
Snyder RJ, Ead J, Glick B, Cuffy C. Dehydrated human amnion/chorion membrane as adjunctive therapy in the multidisciplinary treatment of pyoderma gangrenosum: a case report. Ostomy Wound Manage. 2015;61(9):40–9.
Wirsing PG, Habrom AD, Zehnder TM, Friedli S, Blatti M. Wireless micro current stimulation—an innovative electrical stimulation method for the treatment of patients with leg and diabetic foot ulcers. Int Wound J. 2015;12(6):693–8. https://doi.org/10.1111/iwj.12204.
Pichler M, Larcher L, Holzer M, Exler G, Thuile T, Gatscher B, et al. Surgical treatment of pyoderma gangrenosum with negative pressure wound therapy and split thickness skin grafting under adequate immunosuppression is a valuable treatment option: case series of 15 patients. J Am Acad Dermatol. 2016;74(4):760–5. https://doi.org/10.1016/j.jaad.2015.09.009.
Richmond NA, Lamel SA, Braun LR, Vivas AC, Serena T, Kirsner RS. Epidermal grafting using a novel suction blister-harvesting system for the treatment of pyoderma gangrenosum. JAMA Dermatol. 2014;150(9):999–1000. https://doi.org/10.1001/jamadermatol.2014.1431.
Wollina U. Pyoderma gangrenosum—a review. Orphanet J Rare Dis. 2007;2:19. https://doi.org/10.1186/1750-1172-2-19.
Twillman RK, Long TD, Cathers TA, Mueller DW. Treatment of painful skin ulcers with topical opioids. J Pain Symptom Manag. 1999;17(4):288–92.
Barker S. Analgesic effect of locally applied morphine to pyoderma gangrenosum. Clin Exp Dermatol. 2009;34(1):91–2. https://doi.org/10.1111/j.1365-2230.2008.02912.x.
Beiteke U, Bigge S, Reichenberger C, Gralow I. Pain and pain management in dermatology. J Dtsch Dermatol Ges. 2015;13(10):967–87. https://doi.org/10.1111/ddg.12822.
Herskovitz I, MacQuhae FE, Dickerson JE Jr, Cargill DI, Slade HB, Margolis DJ, et al. Opioids’ effect on healing of venous leg ulcers. J Invest Dermatol. 2017;137(12):2646–9. https://doi.org/10.1016/j.jid.2017.07.837.
Maida V, Corban J. Topical medical cannabis: a new treatment for wound pain-three cases of pyoderma gangrenosum. J Pain Symptom Manag. 2017;54(5):732–6. https://doi.org/10.1016/j.jpainsymman.2017.06.005.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Nutrition, Obesity and Diabetes
Rights and permissions
About this article

Cite this article
McKenzie, F., Arthur, M. & Ortega-Loayza, A.G. Pyoderma Gangrenosum: What Do We Know Now?. Curr Derm Rep 7, 147–157 (2018). https://doi.org/10.1007/s13671-018-0224-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13671-018-0224-y