
Abstract
Purpose of Review
Expanding therapeutic targets from proteins to RNAs opens up new possibilities for neurodegenerative disorders therapeutics development. Recently, a disease-modifying antisense oligonucleotide (ASO) agent was approved for spinal muscular atrophy, suggesting ASOs will fulfill their early promise and become a significant new therapeutic category for neurodegenerative disorders.
Recent Findings
ASOs are in human subjects testing for Huntington disease, monogenic forms of amyotrophic lateral sclerosis, Alzheimer disease, myotonic dystrophy, Leber congenital amaurosis, Usher syndrome, and retinitis pigmentosum, with many more in preclinical development. Current ASO strategies encompass RNA processing modulation, and RNA target breakdown. Broad ASO mechanism categories are protein restoring versus protein lowering. Individual ASO mechanisms of action range from mutation-specific to impacting many proteins.
Summary
Current ASOs show great promise in neurodegenerative disorders. Specific ASO designs and mechanisms may be more tenable in this disease area. Preclinical development is already leveraging early knowledge from these initial clinical trials to develop novel ASO cocktails, new ASO chemical modifications, and new ASO RNA and protein targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Expanding therapeutic targets from proteins to nucleic acids has opened up exciting areas of opportunity in neurodegenerative disorders. Antisense oligonucleotides (ASOs) are synthetic short single-stranded chains of nucleic acids that target native RNAs. An ASO binds to its sense strand target RNA via Watson-Crick base pairing. Proposals of ASOs as potential therapeutics first appeared in the 1970s, with acceleration of research in the 1990s [1, 2, 3•, 4]. New ASO-based therapeutics incorporate advances in synthetic chemistry modifications and understanding of native RNAs. ASOs are now in all stages of therapeutic development [3•, 4].
As ASOs are built from nucleic acids they are genetic therapeutics, although not “gene therapy” as they do not permanently modify native DNA. The terminology is shifting to more clearly classify therapeutics by molecular basis, molecular target, or end mechanism. ASOs are RNA- and DNA-based, and RNA-targeting, and are discussed under end mechanism-based categories such as “protein lowering.”
There is significant past and ongoing pre-clinical ASO research [3•, 5•]; this review focuses on therapeutic compounds tested in humans.
Key Therapeutic Features
ASOs are directly taken up through cell membranes [6, 7]. ASOs do not cross the blood brain barrier, making injection into cerebrospinal fluid (CSF) a necessity for most neurodegenerative disorders [4, 8]. Exceptions discussed below include retinal disorders. ASOs can be highly specific, binding to only one target RNA. They are particularly effective at targeting single-nucleotide polymorphisms (SNPs); examples below also target large RNA repeats.
The oligonucleotide backbone is vulnerable to RNase breakdown. Modifications that reduce ASO degradation, such as first-generation ASO phosphorothioate backbones [2], greatly increase ASOs therapeutic viability. This modification also reduces renal excretion and increases tissue uptake [9••].
Multiple other chemical modifications are used. These broadly include modifications of oligonucleotide backbones, the bases, and sugar moieties, and conjugating oligonucleotides with ligands [5•, 9••]. Modifications enhance ASO stability, target RNA affinity, and target tissue concentrations. Modifications may improve one area, such as tissue targeting, at the expense of another, such as end mechanism. For example, 2′ sugar modifications in second-generation ASOs all confer greater binding affinity, but inhibit RNase H target RNA breakdown. To overcome this, “gapmer” chimera modifications set DNA at the RNase H target area between flanking 2′ modified ribonucleotide zones [9••, 10]. 2,‘4’bridged or locked nucleic acid 2′ modification confers the highest binding affinities but also higher side effect profiles, necessitating further compensatory modifications [9••]. An ASO third generation using nucleic acid analogues is in development [5•].
Chemical modifications, nucleotide sequence design, and RNA target function factor into varying therapeutic strategies. ASOs differ in mechanisms of action at RNA and protein levels. ASOs containing strings of five or more DNA nucleotides can form a DNA-RNA heteroduplex with their target RNA in the nucleus or mRNA in the cytoplasm, triggering RNase H1 degradation. ASOs discussed below use RNase H to lower protein-coding or noncoding RNA targets. ASOs can also promote target RNA degradation through argonaute 2 and RNA interference pathways, or as ribozymes [3•, 9••].
ASOs can alter RNA processing or functions, without target RNA degradation. Noncoding RNAs have numerous potentially manipulable normal functions [11,12,13]. One pre-clinical example comes from spinal muscular atrophy (SMA) research: ASOs disrupt a long chain RNAs normal protein interactions, blocking suppression of SMN2 transcription and potentially boosting functional protein levels [14]. Protein coding RNAs undergo multiple processing steps prior to translation. ASOs can bind to and thus block pre-mRNA splice sites or 5′-cap and 3′-polyadenylation sites [3•]. In examples below, ASOs splice site targeting results in exon insertion, aberrant exon deletion, and exon skipping.
The end results at the protein level include protein reduction, gain of function, or protein restoration. ASOs may increase levels of normal protein, convert severely mutated protein into abnormal but more functional forms, or promote release of toxic RNA-bound proteins. DNA allele-specific and nonspecific strategies are used, with distinct consequences for protein level results and target patient populations.
With their high RNA target specificity, ASOs were initially attractive for monogenetic mendelian neurodegenerative disorders [4]. More recently, clinical trials have expanded into sporadic forms of some disorders (NCT03186989, see below), and general pathological pathways (NCT03815825) [15].
Protein-Coding Pre-mRNA and mRNA Targets
Protein Restoring Strategies
The ASOs in this group address autosomal recessive mutations driving mainly protein loss of function (Table 1). ASOs alter RNA processing in order to restore functional or partially functional protein. Examples here target splice sites. These strategies do not have allele specificity concerns. ASOs may target the mutated RNA transcript itself, or target a different gene product to enhance production of a homologous protein.
Spinal Muscular Atrophy
SMA is an autosomal recessive disorder caused by deletion or mutation of both SMN1 genes, loss of survival motor neuron protein (SMN), and resultant degeneration of spinal cord anterior horn motor neurons [16]. Severity is graded from type 0 (worst) to 5.
In SMA, the ASO target is distinct from both the disease-causal mutation and disease-related gene (Table 2). The nearly identical SMN2 gene usually generates mRNA excluding exon 7, producing non-functioning protein that is rapidly degraded. SMA disease severity is modulated by how many copies of SMN2 the patient has, and what percentage of SMN2-generated mRNA includes exon 7, producing functioning SMN. Nusinersen, an FDA-approved agent, acts on SMN2 pre-mRNA, blocking the intronic splicing silencer that would normally drive exon 7 exclusion. This boosts full length mRNA production. The resulting SMN2-generated SMN protein replaces that lost due to SMN1 mutations. An open-label trial with comparison to longitudinal observational study data in type 1 SMA was the basis for fast-tracking FDA approval [17].
Controversies remain, including application to adult SMA patients and less severe SMA types, whether lifelong therapy is truly required, consequences of gaps in therapy, and the extremely high cost. An observational study of adults with SMA types 2 and 3 receiving nusinersen clinically is ongoing (NCT03709784).
Inherited Retinal Disorders
The eye as an ASO target area has multiple advantages [18•]. These include ease of local administration, target coverage especially compared to whole central nervous system (CNS), and natural limits on systemic side effects. ASOs can be delivered via repeated intravitreal injections, and are well tolerated [19]. The first approved ASO therapeutic addressed cytomegalovirus retinitis in immunocompromised patients [20, 21].
Inherited retinal dystrophies (IRDs) form a large heterogeneous group of rare neurodegenerative diseases [22]. Many are caused by mutations affecting RNA splicing, making splicing-modulating ASOs natural therapeutic fits [23, 24]. Unlike nusinersen, which restores an exon to create normal protein, strategies discussed below cut out aberrant or mutated exons (Table 2).
A phase 2/3 randomized controlled dose finding study (NCT03913143) in the primary ciliopathy form of Leber congenital amaurosis follows a completed phase 1/2 open-label dose escalation (NCT03140969). In this childhood blindness, an intronic mutation in the centrosomal protein 290 (CEP290) gene creates a strong splice donor site, driving aberrant pre-mRNA splicing and insertion of a cryptic exon with a premature stop codon [25]. Abnormal mRNA transcripts are degraded, or create truncated inactive protein. QR-110 blocks the aberrant splice site, preventing cryptic exon insertion and increasing normal mRNA and CEP290 protein levels [26, 27••]. QR-110 is the only directly mutation targeted ASO in this group.
In the phase 1/2 trial, QR-110 intravitreal injections were done in one eye every 3 months. Early analyses of results from ten participants were triggered by one very strong responder. In addition to excellent safety data, improvements in visual acuity and full-field stimulus testing were observed in a subgroup 3 months post initial injections, even excluding the one strong responder [27••], although small open-label study results must be interpreted with caution. The phase 2/3 study projected enrollment is 30. An open-label extension study is ongoing (NCT03913130).
Retinitis pigmentosa (RP), progressive degeneration of photoreceptors causing vision loss, occurs in isolation (nonsyndromic, NSRP) or as part of larger syndromes. Usher syndrome is a genetic disorder family with inner ear hair cell and retinal photoreceptor degeneration causing deaf-blindness [28]. Much ASO pre-clinical work focuses on Usher syndrome hearing loss [29•]. Current clinical trial work addresses the RP component. Mutations in USH2A, encoding usherin, cause autosomal recessive NSRP, and are a common cause of Usher syndrome type 2 [30]. There are nearly 200 reported disease-causing USH2A mutations. Mutations in exon 13, including premature stop codons and frameshifts, are the most common in non-Finnish European descent populations [31]. Using ASOs to block specific pre-mRNA splice sites and skip usherin exon 13 maintains the normal open reading frame, changing abnormal usherin from severely truncated to a nontruncated protein missing one exon, in theory greatly mitigating mutation impacts, although the resulting protein is not identical to wildtype usherin. A similar strategy is in development for Duchenne muscular dystrophy [32••]. The exon 13 skipping ASO QR-421a is in active phase 1/2 clinical trial (NCT03780257) [29•].
Protein Lowering Strategies
A common ASO strategy is to utilize native RNA breakdown pathways to lower specific mRNA and thus specific protein levels. Two sets of ASOs discussed below are allele-specific, targeting only the mutated RNA through different methods. Others are not allele specific: both mutated and wildtype mRNAs are lowered. The various approaches have distinct pros and cons (Table 1). Several similar ASO strategies are in preclinical development, such as spinocerebellar ataxia and ALS-associated ATXN2 CAG repeat expansions [4, 33].
Amyotrophic Lateral Sclerosis
Therapeutic ASOs in active development target mutations that cause amyotrophic lateral sclerosis (ALS) or varied motor neuron disease (MND)/frontotemporal dementia (FTD) phenotypes (Table 2). Roughly 10% of ALS, the most common MND, is monogenetic (OMIM 105400). The second most common monogenetic ALS, after C9orf72 mutations, is SOD1 mutations [34]. In addition to agents detailed below, multiple ASOs are in preclinical development [35•].
SOD1 mutations represent about 20% of monogenetic ALS [36]. Autosomal dominant mutations are thought to act through a toxic gain in function; thus, current ASOs promote RNAse H1 pre-mRNA breakdown and superoxide dismutase (SOD1) protein lowering. The first phase 1 trial (NCT01041222) for an intrathecally delivered SOD1-lowering ASO (ASO 333611) yielded encouraging tolerance and feasibility data [37]. Screening of next-generation ASOs from the same developers identified multiple candidates with greater potency for SOD1 lowering than 333611 and extended survival in animal models [38]. One of the newer ASOs, BIIB067 (tofersen), completed phase 1/2 of a three-part single- and multiple-ascending dose study in 2019. Results, presented in abstract form, included statistically significant reduction in CSF SOD1 in the highest dose cohort (10 participants) versus placebo (12 participants), and some slowing of change in exploratory functional and clinical outcomes [39]. The part C phase 3 randomized placebo-controlled trial section is now open (NCT02623699). There is also an ongoing open-label extension (NCT03070119).
These trials only enroll confirmed SOD1 mutation ALS (SOD1-ALS) patients; however, the ASOs are SOD1-specific, not mutation specific. What role SOD1 plays in sporadic ALS, and whether SOD1 lowering in sporadic MNDs is beneficial, is unknown. These agents are also allele non-specific: both mutated and wildtype SOD1 proteins decrease. Balancing lowering mutated protein as much as possible against potential negatives of lowering wildtype protein is a common theme in this ASO therapeutic area.
Tauopathies
The microtubule associated protein tau is the pathogenic hallmark of multiple neurodegenerative disorders, including progressive supranuclear palsy and corticobasal degeneration; MAPT mutations cause forms of frontotemporal dementia [40]. MAPT variants are genetic risk factors in disorders such as Parkinson disease [41]. Pre-clinical work on MAPT-related ASOs comprises several strategies [42], reflecting the variety of protein aggregate pathology in sporadic disease and disease-causing MAPT mutations. While BIIB080 was tested in animal models against mutant human MAPT [43], this ASO is allele non-specific, reducing all forms of MAPT mRNA and tau protein. A phase 1/2 randomized controlled trial of BIIB080 in early Alzheimer disease (AD) is ongoing (NCT03186989). In contrast to ASO ALS trials, this study is in sporadic disease (Table 2). Unknowns include the negative impact of lowering all tau protein isoforms, impact of protein lowering in a sporadic disorder, and impact of addressing one pathologic protein in a multi-protein disorder. AD pathology features both tau aggregates and amyloid beta deposits; tau aggregates occur earlier [44, 45]. In theory reducing normal tau could also reduce AD amyloid pathology, although their relationship is not fully understood [40].
Huntington Disease
Huntington disease (HD) is a monogenetic neurodegenerative disorder caused by a CAG expansion in exon 1 of the huntingtin (HTT) gene. There are both allele-specific and non-specific ASO strategies in active clinical trial testing, all huntingtin protein lowering (Table 2).
There is an ongoing pivotal phase 3 trial of the non-allele specific RO7234292 (tominersen) (NCT03761849). Intrathecal RO7234292 injections are done every 8 or 16 weeks. The same compound completed phase 1/2 (NCT02519036) with open label extension (NCT03342053). RO7234292 was well tolerated in 46 patients with clinically manifest HD. Dose-dependent CSF mutant huntingtin reductions were observed [46••]. This exciting result set the stage for the pivotal trial, although it is unclear how CSF protein levels will correlate with impact on human disease. Data on potential biomarkers from phase 1/2 trials are being compared to those in an ongoing prospective natural history study (NCT03664804) in an attempt to clarify biomarkers for disease progression and ASO efficacy.
RO7234292 lowers both mutated and wildtype protein. The unknown safe level for lowering wildtype huntingtin puts a limit on how low this or similar allele-nonspecific therapeutics can drive down target protein. Huntingtin is a very large protein with numerous proposed normal functions [47, 48]. Potential neurodevelopmental roles suggest loss of function mutation effects, and possible protective effects of wildtype protein against mutant huntingtin [49•]. Conversely, the normal allele size does not influence onset age of motor signs [50, 51]. Homozygote HD cases are rare, limiting information on the impact of naturally producing no normally sized huntingtin. Whether homozygotes have a more severe clinical phenotype than heterozygotes is unclear [52,53,54].
Allele-specific strategies leave wildtype RNA and protein intact. Challenges for these ASOs depend on the mechanism of allele specificity. A pair of ongoing phase 1/2 clinical trials in HD (NCT03225833, NCT03225846) represent a distinct ASO-based protein-lowering strategy: allele-specific haplotype targeting. Patterns of genetic polymorphisms comprising HTT haplotype backgrounds occur with different frequencies in different populations [55,56,57]. Targeting a haplotype-defining SNP cis to HTT generates allele-specific protein lowering if the patient is heterozygous for the SNP on the mutated HTT allele [58, 59]. Some common haplotypes such as A1 and A2 are often associated with expanded CAG repeat HTT mutations rather than normal CAG repeat lengths [57, 58]. At a population level, higher HD prevalence appears tied to the presence of these higher risk haplotypes, with many patients of European or mixed ancestry and some Latin American populations sharing a common remote original mutation; in contrast, low HD prevalence populations have rarer multiple independent CAG expansion mutations on a mix of other HTT haplotypes [47, 51, 57, 60, 61]. The current allele-specific HD ASOs leverage the relatively high general and mutation-specific prevalence of some HTT haplotypes. WVE-120101 is targeted to the SNP rs362307 (SNP1), a defining A1 haplotype marker common in some European populations [59]. WVE-120102 targets rs362331 (SNP2) which distinguishes A and B haplogroups from C [59]. Exact percentages of HD patients these ASOs could impact is unclear but is at least population dependent. A study of Canadian, Swiss, Italian, and French population data estimated a maximum 80% coverage in those specific HD populations if A1, A2, and A3a haplotypes were all targeted [59].
Haplotype targeting presents a unique challenge. The number of patients potentially benefiting per ASO depends on haplotype background population frequencies, plus whether patients with targeted haplotypes are heterozygous for the haplotype-defining SNP on the CAG-expanded HTT allele. New ASOs will have to be developed to even partially cover non-Northern European HD populations [62••]. As examples, A1 and A2 haplotypes are absent or rare in most east Asian general populations and black South Africans; HD causal mutations are instead mainly observed on C and B haplotypes respectively [56, 61, 63].
The production platform for WVE-120101 and WVE-120102 differs from that of other ASOs. The chirality of each backbone phosphorothioate bond is specified, creating a “stereopure” agent, in contrast to all of the other ASOs presented in this review, which are stereoisomeric mixtures. Both approaches have their own pros and cons [64]. Using stereopure ASOs may improve RNAse H1 degradation of target RNAs [65].
Autosomal Dominant Retinitis Pigmentosa
The allele-specific QR113 ASO targets the dominant negative P23H rhodopsin mutation [66]. First described in an Irish origin pedigree, this coding region point mutation causes the most common autosomal dominant NSRP in the USA [67], although it is rare in other populations [68]. A phase 1/2 trial (NCT04123626) is ongoing.
QR113 is mutation-specific: the P23H mutation itself provides an allele-specific ASO target [69]. Advantages include no impact on wildtype rhodopsin protein levels, and no haplotype-targeting genetic subpopulation issues. However, QR113 will not impact other RPs, including those from other rhodopsin mutations.
Noncoding RNA Targets
There are several types of noncoding RNAs with highly varied functions. In some repeat expansion disorders, the disease pathology is not fully understood, but toxic noncoding RNAs (lncRNAs) are thought to play a key role [70••, 71]. In these autosomal dominant toxic RNA gain of function disorders, the genetic mutation generates RNA transcripts or transcript fragments which exert disease-causing effects. ASOs can easily access nuclear-based toxic lncRNAs and any RNA byproducts in cytoplasm. The expanded repeat RNAs may generate toxic dipeptides through repeat-associated non-ATG (RAN) translation, or have direct negative effects on cellular pathways. ASOs using RNase H1 breakdown focus on lowering toxic RNA transcripts and associated RAN-generated products. ASOs can also be designed to directly disrupt toxic lncRNA-protein binding [9••]. Pre-clinical work is also exploring how ASO therapeutics could leverage normal lncRNA functions [14, 70••].
In the disorders discussed below, the mutated allele produces less mRNA and protein, with loss of function consequences. ASOs designs must take this into account. Another shared challenge is the difficulty of adequately impacting a large toxic RNA, compared to the SNP-based ASO mechanisms discussed above.
Myotonic Dystrophy
Myotonic dystrophy is caused by greatly expanded 3′UTR CTG or CCTG repeats in one of two genes [72, 73]. The massively expanded CUG or CCUG RNA transcripts section has multiple toxic effects [71]. The expanded repeat lncRNA binds RNA splicing proteins into nuclear foci, rendering the sequestered proteins non-functional and secondarily disrupting normal splicing and activity of many other proteins. Reducing foci formation or releasing proteins from dynamic foci directly restores sequestered proteins, and indirectly restores functional levels of many more proteins by normalizing RNA splicing. The expanded lncRNAs can also negatively impact cell signaling, and may generate toxic dipeptides through RAN translation. All of these potential pathological mechanisms may contribute to disease. There is also evidence that disease-causal mutations suppress mutant allele expression, creating loss of normal disease-gene generated protein. This could be ameliorated or exacerbated by ASOs with unclear downstream consequences [74••].
ASOs for myotonic dystrophy type 1 (DM1) (OMIM 160900) are much farther along in development than those for type 2 [74••]. ASO development to date focuses on skeletal muscles: DM1 is the most common adult onset muscular dystrophy, causing weakness, muscle atrophy, and myotonia [72, 73]. This focus includes systemic delivery methods, muscle function efficacy endpoints, and muscle biopsy-based biomarkers. DM1 is caused by a 3′ untranslated region CTG triplet repeat expansion in the gene encoding dystrophin myotonica protein kinase (DMPK) [75, 76]. ASOs for DM1 focus on RNase H1 breakdown of the toxic RNA transcript, and releasing of sequestered proteins [77, 78]. The IONIS-DMPK-2.5Rx ASO halted development after a phase 1/2a multiple dose ascending clinical trial (NCT02312011). Although safety and tolerability endpoints were encouraging, and small trends in biomarkers were reported, muscle biopsies showed low target tissue ASO levels [79].
The trial results highlight continued needs common across ASOs: increased potency and improved ASO levels in target tissues. Development of both RNA-degrading and RNA-protein-binding steric blocker ASOs for DM1 continues, although all agents remain far from clinical trials. Preclinical work highlights challenges in ASO backbone modification, tissue targeting, and ASO engagement with very large RNA repeat expansion areas versus SNPs [9••, 80,81,82,83].
In addition to muscular dystrophy, DM1 causes early onset cataracts and cardiac dysrhythmias; other features such as insulin resistance and hypogonadism are common [73]. Myotonic dystrophy has also long been recognized as a widespread nervous system disorder [84]. DM1 causes significant neuropsychiatric impairment due to underlying global cortical neurodegeneration [73, 85]. Addressing DM1 neurodegeneration beyond muscle atrophy will require different approaches than those tested to date. For example, subcutaneously administered ASOs will not cross the blood brain barrier.
Motor Neuron Disease and Frontotemporal Dementia
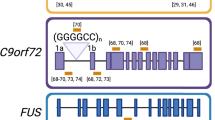
ALS shares some monogenetic causes with frontotemporal dementia (FTD) [86, 87]. Hexanucleotide G4C2 repeat expansion (HRE) mutations in intron 1 of the chromosome 9 open reading frame 72 gene (C9orf72) [88, 89] account for over 30% of monogenetic ALS, and cause MND, FTD, mixed MND/FTD, and other neurodegenerative phenotypes [86, 90, 91].
There are multiple non-exclusive proposed pathogenic mechanisms in HRE C9orf72 disease [92, 93]. As discussed above in DM1, highly expanded lncRNAs can sequester RNA-binding proteins into nuclear foci. Both antisense (G2C4) and sense (G4C2) expanded C9orf7 RNA transcripts have been reported in nuclear foci, and both may undergo RAN translation to create toxic dipeptide repeat proteins. ASOs usually target sense transcripts; strategies that also cover HRE C9orf7 antisense transcripts may be required [35•]. The HRE is in the open reading frame in intron 1, and is transcribed into some of the normal pre-mRNA transcripts. C9orf72 protein derived from the mutant allele is reduced, which can cause loss of function pathology. Current ASO designs often target areas 5′ to the HRE to avoid worsening reductions in C9orf7 mRNA and protein [35•, 93, 94].
There is an ongoing multi-cohort phase 1 study of BIIB078 (IONIS-C9Rx) in C9orf72-ALS (NCT03626012). This ASO creates RNase H1-based reduction in toxic C9orf72 RNA. It is targeted upstream of exon 1b. The strategy covers some but not all of the proposed HRE pathogenic mechanisms. In animal models, similar ASOs decreased sense lncRNA-based nuclear foci and dipeptide repeat proteins, without worsening the decrease in exon1b-containing C9orf72 mRNA, but also without reducing antisense lncRNA-based nuclear foci [95].
Shared Challenges
Toxic Effects
Potential toxic effects specific to individual ASO targets and strategies are covered above. Off-target binding is also possible. One key example is how an RNase-activating ASO could lower unintended targets [4, 9••]. ASOs can also interact with proteins, causing toxic immune responses, thrombocytopenia, and end organ damage [9••, 96]. Different chemical modifications confer different toxicologic profiles, which may need to be countered by further modifications [9••, 97]. In clinical trials of inotersen for hereditary transthyretin amyloidosis polyneuropathy (NCT01737398), cases of severe thrombocytopenia and glomerulonephritis were reported, along with overall lowered platelet counts in the ASO-treated group [3•]. Thrombocytopenia may be related to the type of 2′ ASO modification used [98].
Delivery to Affected Tissues
ASOs are all eventually broken down, making their effects temporary [9••]. Debate remains over clearance time, duration of effects particularly in CNS, and thus frequency of dosing, but repeated dosing will be required to maintain benefit for all the ASOs presented above [3•]. For retinal disorders, single subretinal injections of adeno-associated viral vector-containing ASOs are possible [19]. The impermanence of ASO effects may be an advantage in terms of reversing any side effects, or allowing patients to shift to other future treatments. Repeated intrathecal and intravitreal injections appear well tolerated [19, 99]. Consequences of gaps in treatment are not yet understood. Potential issues include supply chain disruptions and varying regional availability of injection providers.
Repeated lumbar intrathecal injections are the main ASO delivery for neurodegenerative disorders. This may be advantageous for spinal cord or cortically based pathology, and less effective in disorders biased to deep brain structures [7, 17], although there is evidence for partial, potentially adequate ASO penetration of whole brain after lumbar intrathecal injection [64, 99]. Alternatives such as intraventricular or combined CSF infusion site strategies are not in current testing [100].
Delivery to affected tissues beyond CNS is also hampered by the inability to directly flood all affected tissue with ASOs. For example, muscles are found throughout the body; muscle fibers are within bundles of connective tissue. Chemical modifications can increase ASO potency by improving tissue delivery. Conjugated ASOs can be designed to target striated muscle [81] or pancreatic cells [101]. GalNAc-conjugated ASOs targeting hepatocytes are in active trials [102, 103].
Impact on Disease Pathology
These therapeutics are disease-modifying, attempting to slow disease progression, a significant advance particularly in neurodegenerative disorders. Some ASOs will only partially impact disease pathology by design. Examples are lowering tau in early AD, and using exon skipping to produce a less abnormal protein. Successfully addressing one disease process, such as toxic lncRNA levels, may exacerbate another, such as loss of normal protein. ASO targeting may not cover all of the disease-contributing RNA and protein products, as in toxic lncRNA target examples. This issue can also occur in non-lncRNA-based disease. For example, mutant HTT pre-mRNA may be aberrantly spliced, generating CAG repeat-containing mRNA and pathogenic exon 1 protein fragments not covered by ASOs targeting more 5′ HTT RNA areas [64, 104].
Even for ASOs fully addressing all “upstream” RNA products, within each cell proteins will not be fully restored or lowered. ASO impact within cells may depend on single target SNP vs multitarget per big lncRNA [81]. The level of benefit for partially impacting protein levels ranges from excellent to unclear. Strategies that restore one key protein (type 1 SMA) may have more impact than secondarily restoring many proteins (DM1), or protein-lowering strategies. An additional concern for non-allele specific protein lowering agents and protein-lowering ASOs in sporadic disease is safety of lowering wildtype proteins. This is challenging to effectively model in pre-clinical experiments, may vary widely across proteins, and may vary within an individual’s lifespan and disease course.
Translating Trial Results to Clinical Use
Early intervention is considered important as neurodegeneration is irreversible. Mutation-driven disorders have well documented pathology long prior to clear clinical signs. Toxic RNA-sequestered protein foci are observed throughout development in DM1, a generally adult onset disorder [105]. Early may be relatively easy to define: SMA is often diagnosed in early childhood. Some very early testing of nusinersen is underway [106]. For highly variable or late onset disorders, tradeoffs in repeated therapeutic administration against modifying disease curve will be more difficult to discern. HD has a monogenic easily testable cause, a wide age of onset range with nonmanifest, prodromal, and clinically motor manifest stages, and promising biomarkers that may predict clinical outcomes [107•]. At the same time, there are controversies over how to include cognitive symptoms in clinical definitions and trial outcomes, and huntingtin lowering may have particularly negative consequences during neurodevelopment. It remains unclear when and how to test and then deploy ASOs in nonmanifest patient populations, whether young HD mutation carriers or unaffected older adults with sporadic AD risks.
Phenotypic variability, how to time effective interventions, biomarker use, and length of time needed to adequately observe therapeutic impacts are some challenges in clinical trials design for neurodegenerative disorders. Creative solutions include the SMA trial using comparison to observational study data as a way to accelerate ASO development. Other issues are more specific to ASOs. Some stem from ASOs strength, their ability to target SNPs. For an available ASO, who will pay for the type of genetic testing required to determine genetic subpopulations? Also, no single ASO will cover all patients if there are many different mutations (ALS, IRDs) or haplotype ASO targets (HD) within the same disease. One potential solution is testing ASO cocktails that address multiple genetic subpopulations with one therapeutic compound [108•].
Starting an ASOs development in disease subpopulations can help create relatively homogeneous clinical trial patient populations. More phenotypically homogeneous populations confer power and analysis advantages, but narrow clinical trial populations increase difficulties translating results into clinical use. Outside of scientific considerations, regulators or insurance agencies may demand treated patients also fit into narrow disease subpopulations to access coverage. This is already playing out in SMA. A single mutation can still produce wide phenotypic variability, one reason initial HD ASO studies focused on narrowly defined early manifest HD populations [109]. A current pivotal trial expanded this to early and moderately affected HD patients (NCT03761849). This still leaves future application in nonmanifest HD mutation carriers unclear. ALS provides a genetic subpopulation example: in an observational study A4V SOD1 was more homogeneous, and severe, than other SOD1-ALS [110]. Current trials are enrolling all SOD1-ALS. This potentially lowers power and slows trial completion, but eases results interpretation for all SOD1-ALS; translating use to sporadic ALS and to SOD1 mutation carriers is the next debate.
If an initial ASO is approved, must every similar ASO undergo the same safety and efficacy testing? How is “similar” best defined? Ongoing conversations with regulators will need to parallel scientific research [108•]. Who will develop ASOs for very rare mutations? For some disorders, these access questions intersect with larger health disparities: will haplotypes (HD [62••]) or mutations (NSRP [67, 68]) concentrated in non-Northern European or US minority groups lag in ASO therapeutic development and access? How can this possibility be mitigated when ASO development is currently focused in the USA and Europe?
ALS again provides an example. Fused in sarcoma gene (FUS) mutations overall account for only 5% of familial and 1% of sporadic ALS [111]. The P525L FUS mutation is a rare cause of juvenile ALS [112]. Recently, one P525L FUS-ALS patient, her treating ALS clinical research group, and the FDA collaborated to accelerate development of a P525L-targeted ASO (jacifusen) [113] which drives allele-specific mutated FUS RNA and protein lowering. The ASO’s namesake passed away in March 2020 after twelve monthly intrathecal infusions [114]. The ALS Association and Project ALS have funded jacifusen testing in eight more P525L FUS-ALS patients [115]. This work directly impacts a very small number of patients, but could help streamline development, testing and approval of novel ASOs also targeting rare mutations [116].
Partnerships between patients, researchers, foundations, and government agencies are one method for increasing therapeutic access across the full patient range. Accumulated safety and efficacy data may obviate the need for expensive extensive testing of every ASO covering a specific genetic target, such as FUS or rhodopsin SNP mutations, or rare HTT haplotypes. Some categories, such as allele-nonspecific protein lowering (tau) or mutations causing the same disease phenotype (FUS vs SOD1 ALS), may be able to shorten safety testing and focus pivotal trials on efficacy.
Future Directions
ASO therapeutic development is already expanding from monogenic neurodegenerative disorders to sporadic disorders. Current ASOs focus on mRNA and lncRNA targets. Future targets include micro RNAs [3•, 117•].
The range of ASO therapeutic strategies continues to grow. Cocktails could use multiple ASO strategies to amplify one end result. Preclinical work in SMA uses SMN2 splicing ASOs similar to nusinersin along with an ASO to degrade an lncRNA that normally downregulates SMN expression [70••, 118], increasing SMN through multiple pathways. Future therapeutics may act more indirectly on disease pathways. An example is ASO-based ataxin-2 lowering. Transactive response DNA-binding protein 43 kDa (TDP-43) aggregates are a common pathology across nearly all ALS forms. Lowering ataxin-2 via ASOs in animal models lowers TDP-43 aggregation and decreases neurodegeneration [119, 120]. Finally, ASOs can address downstream neuropathology. In contrast to IRDs, age-related macular degeneration (AMD) is a common, multifactorial disorder with several genetic and non-genetic risk factors driving eventual loss of macular retinal pigmented epithelium and photoreceptors [15]. IONIS-FB-LRx in development for geographic atrophy in AMD targets complement pathways (NCT03815825). The same ASO is in clinical trials for IgA nephropathy (NCT04014335). These general effect ASOs could be paired with disease- or mutation-specific genetic therapeutics. Conversely, disease-modifying ASOs could be paired with non-ASO therapeutics that address more downstream pathology, such as neuroinflammation.
Neurodegenerative disorders are often multiorgan syndromes. The need to target CNS, skeletal muscle, and other tissues in DM1 is an example. Classic CNS disorders such as ALS and HD can be thought of as systemic disorders [93, 121]. Future combination therapeutics may utilize sets of ASOs with tissue-specific chemical modifications and different routes of administration to cover both CNS and non-CNS tissues.
Conclusions
ASOs radically expand the reach of disease-modifying therapeutics in neurodegenerative disorders. Although ASOs have specific intrinsic challenges, this remains a rapidly expanding, very promising clinical trial field. Many advances are on the horizon, from third-generation ASOs to novel therapeutic targets to ASO cocktail agents. ASOs will remain a key component of the next wave of neurodegenerative disorder therapeutics.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Crooke ST, Lemonidis KM, Neilson L, Griffey R, Lesnik EA, Monia BP. Kinetic characteristics of Escherichia coli RNase H1: cleavage of various antisense oligonucleotide-RNA duplexes. Biochem J. 1995;312(Pt 2):599–608. https://doi.org/10.1042/bj3120599.
Gao WY, Han FS, Storm C, Egan W, Cheng YC. Phosphorothioate oligonucleotides are inhibitors of human DNA polymerases and RNase H: implications for antisense technology. Mol Pharmacol. 1992;41(2):223–9.
• Bennett CF. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med. 2019;70:307–21. https://doi.org/10.1146/annurev-med-041217-010829Overview of ASO therapeutic strategies and pharmacology focused on clinically available agents.
Scoles DR, Pulst SM. Oligonucleotide therapeutics in neurodegenerative diseases. RNA Biol. 2018;15(6):707–14. https://doi.org/10.1080/15476286.2018.1454812.
• Egli M, Manoharan M. Re-engineering RNA molecules into therapeutic agents. Acc Chem Res. 2019;52(4):1036–47. https://doi.org/10.1021/acs.accounts.8b00650Detailed look at chemical modifications and their impact on nucleic acids as therapeutics.
Crooke ST, Wang S, Vickers TA, Shen W, Liang XH. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. 2017;35(3):230–7. https://doi.org/10.1038/nbt.3779.
Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51. https://doi.org/10.1016/j.addr.2015.01.008.
Rigo F, Chun SJ, Norris DA, Hung G, Lee S, Matson J, et al. Pharmacology of a central nervous system delivered 2'-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther. 2014;350(1):46–55. https://doi.org/10.1124/jpet.113.212407.
•• Bennett CF, Baker BF, Pham N, Swayze E, Geary RS. Pharmacology of Antisense Drugs. Annu Rev Pharmacol Toxicol. 2017;57:81–105. https://doi.org/10.1146/annurev-pharmtox-010716-104846Detailed review of ASO chemical modifications, pharmacokinetics, toxicology.
Monia BP, Lesnik EA, Gonzalez C, Lima WF, McGee D, Guinosso CJ, et al. Evaluation of 2′-modified oligonucleotides containing 2′-deoxy gaps as antisense inhibitors of gene expression. J Biol Chem. 1993;268(19):14514–22.
Sharp PA. The centrality of RNA. Cell. 2009;136(4):577–80. https://doi.org/10.1016/j.cell.2009.02.007.
Goff LA, Rinn JL. Linking RNA biology to lncRNAs. Genome Res. 2015;25(10):1456–65. https://doi.org/10.1101/gr.191122.115.
Bartel DP. Metazoan MicroRNAs. Cell. 2018;173(1):20–51. https://doi.org/10.1016/j.cell.2018.03.006.
Woo CJ, Maier VK, Davey R, Brennan J, Li G, Brothers J 2nd, et al. Gene activation of SMN by selective disruption of lncRNA-mediated recruitment of PRC2 for the treatment of spinal muscular atrophy. Proc Natl Acad Sci U S A. 2017;114(8):E1509–E18. https://doi.org/10.1073/pnas.1616521114.
Wu J, Sun X. Complement system and age-related macular degeneration: drugs and challenges. Drug Des Devel Ther. 2019;13:2413–25. https://doi.org/10.2147/DDDT.S206355.
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–65. https://doi.org/10.1016/0092-8674(95)90460-3.
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–26. https://doi.org/10.1016/S0140-6736(16)31408-8.
• Vazquez-Dominguez I, Garanto A, Collin RWJ. Molecular therapies for inherited retinal diseases-current standing, opportunities and challenges. Genes (Basel). 2019;10(9). https://doi.org/10.3390/genes10090654Overview of a key growing area in molecular therapeutics for neurodegenerative disorders.
Rowe-Rendleman CL, Durazo SA, Kompella UB, Rittenhouse KD, Di Polo A, Weiner AL, et al. Drug and gene delivery to the back of the eye: from bench to bedside. Invest Ophthalmol Vis Sci. 2014;55(4):2714–30. https://doi.org/10.1167/iovs.13-13707.
Vitravene Study G. Safety of intravitreous fomivirsen for treatment of cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol. 2002;133(4):484–98. https://doi.org/10.1016/s0002-9394(02)01332-6.
Vitravene Study G. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol. 2002;133(4):467–74. https://doi.org/10.1016/s0002-9394(02)01327-2.
Khan M, Fadaie Z, Cornelis SS, Cremers FPM, Roosing S. Identification and analysis of genes associated with inherited retinal diseases. Methods Mol Biol. 1834;2019:3–27. https://doi.org/10.1007/978-1-4939-8669-9_1.
Hammond SM, Wood MJ. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011;27(5):196–205. https://doi.org/10.1016/j.tig.2011.02.004.
Bacchi N, Casarosa S, Denti MA. Splicing-correcting therapeutic approaches for retinal dystrophies: where endogenous gene regulation and specificity matter. Invest Ophthalmol Vis Sci. 2014;55(5):3285–94. https://doi.org/10.1167/iovs.14-14544.
den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79(3):556–61. https://doi.org/10.1086/507318.
Dulla K, Aguila M, Lane A, Jovanovic K, Parfitt DA, Schulkens I, et al. Splice-modulating oligonucleotide QR-110 restores CEP290 mRNA and function in human c.2991+1655A>G LCA10 models. Mol Ther Nucleic Acids. 2018;12:730–40. https://doi.org/10.1016/j.omtn.2018.07.010.
•• Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med. 2019;25(2):225–8. https://doi.org/10.1038/s41591-018-0295-0Although this is a small open label study, it is one of very few peer-reviewed publications of clinical trial data in this field.
Mathur P, Yang J. Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta. 2015;1852(3):406–20. https://doi.org/10.1016/j.bbadis.2014.11.020.
• Hastings ML, Jones TA. Antisense Oligonucleotides for the Treatment of Inner Ear Dysfunction. Neurotherapeutics. 2019;16(2):348–59. https://doi.org/10.1007/s13311-019-00729-0Overview of pros and cons of ASO therapeutics in a distinct area of neurological disease.
Yan D, Liu XZ. Genetics and pathological mechanisms of usher syndrome. J Hum Genet. 2010;55(6):327–35. https://doi.org/10.1038/jhg.2010.29.
Yan D, Ouyang X, Patterson DM, Du LL, Jacobson SG, Liu XZ. Mutation analysis in the long isoform of USH2A in American patients with usher syndrome type II. J Hum Genet. 2009;54(12):732–8. https://doi.org/10.1038/jhg.2009.107.
•• Li D, Mastaglia FL, Fletcher S, Wilton SD. Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol Sci. 2018;39(11):982–94. https://doi.org/10.1016/j.tips.2018.09.001.detailsWork on a specific ASO strategy relevant to neurodegenerative and other neurological disorders.
Al-Chalabi A, Durr A, Wood NW, Parkinson MH, Camuzat A, Hulot JS, et al. Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS One. 2009;4(9):e7114. https://doi.org/10.1371/journal.pone.0007114.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62.
• Ly CV, Miller TM. Emerging antisense oligonucleotide and viral therapies for amyotrophic lateral sclerosis. Curr Opin Neurol. 2018;31(5):648–54. https://doi.org/10.1097/WCO.0000000000000594Covers clinical and pre-clinical work relevant to ALS and other neurodegenerative disorders.
Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 1607;2015:75–93. https://doi.org/10.1016/j.brainres.2014.10.009.
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–42. https://doi.org/10.1016/S1474-4422(13)70061-9.
McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest. 2018;128(8):3558–67. https://doi.org/10.1172/JCI99081.
Miller TM, Cudkowicz ME, Shaw PJ, Graham D, Fradette S, Houshyar H, et al. Safety, PK, PD, and exploratory efficacy in a single and multiple-dose study of a SOD1 antisense oligonucleotide (BIIB067) administered to participants with ALS. Neurology. 2019;92(15 Supplement):Emerging Science Abstracts. https://doi.org/10.1212/WNL.0000000000007887.
Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12(6):609–22. https://doi.org/10.1016/S1474-4422(13)70090-5.
Zhang CC, Zhu JX, Wan Y, Tan L, Wang HF, Yu JT, et al. Meta-analysis of the association between variants in MAPT and neurodegenerative diseases. Oncotarget. 2017;8(27):44994–5007. https://doi.org/10.18632/oncotarget.16690.
Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, Wozniak DF, et al. Increased 4R-tau induces pathological changes in a human-tau mouse model. Neuron. 2016;90(5):941–7. https://doi.org/10.1016/j.neuron.2016.04.042.
DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374). https://doi.org/10.1126/scitranslmed.aag0481.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–59. https://doi.org/10.1007/BF00308809.
Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960–9. https://doi.org/10.1097/NEN.0b013e318232a379.
•• Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, et al. Targeting Huntingtin expression in patients with Huntington’s disease. N Engl J Med. 2019;380(24):2307–16. https://doi.org/10.1056/NEJMoa1900907Peer reviewed report of initial clinical trial data for an ASO in a CNS-based neurodegenerative disease.
Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005. https://doi.org/10.1038/nrdp.2015.5.
Saudou F, Humbert S. The biology of Huntingtin. Neuron. 2016;89(5):910–26. https://doi.org/10.1016/j.neuron.2016.02.003.
• Wiatr K, Szlachcic WJ, Trzeciak M, Figlerowicz M, Figiel M. Huntington Disease as a neurodevelopmental disorder and early Signs of the Disease in Stem Cells. Mol Neurobiol. 2018;55(4):3351–71. https://doi.org/10.1007/s12035-017-0477-7Important concepts to consider in non-allele specific protein lowering therapeutic strategies, even for apparently straightforward autosomal dominant mutations.
Lee JM, Ramos EM, Lee JH, Gillis T, Mysore JS, Hayden MR, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78(10):690–5. https://doi.org/10.1212/WNL.0b013e318249f683.
Gusella JF, MacDonald ME, Lee JM. Genetic modifiers of Huntington’s disease. Mov Disord. 2014;29(11):1359–65. https://doi.org/10.1002/mds.26001.
Squitieri F, Gellera C, Cannella M, Mariotti C, Cislaghi G, Rubinsztein DC, et al. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain. 2003;126(Pt 4):946–55. https://doi.org/10.1093/brain/awg077.
Ganesh A, Galetta S. Editors’ note: clinical manifestations of homozygote allele carriers in Huntington disease. Neurology. 2020;94(16):722. https://doi.org/10.1212/WNL.0000000000009305.
Cubo E, Martinez-Horta SI, Santalo FS, Descalls AM, Calvo S, Gil-Polo C, et al. Clinical manifestations of homozygote allele carriers in Huntington disease. Neurology. 2019;92(18):e2101–e8. https://doi.org/10.1212/WNL.0000000000007147.
Squitieri F, Andrew SE, Goldberg YP, Kremer B, Spence N, Zeisler J, et al. DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence. Hum Mol Genet. 1994;3(12):2103–14.
Warby SC, Visscher H, Collins JA, Doty CN, Carter C, Butland SL, et al. HTT haplotypes contribute to differences in Huntington disease prevalence between Europe and East Asia. Eur J Hum Genet. 2011;19(5):561–6. https://doi.org/10.1038/ejhg.2010.229.
Kay C, Hayden MR, Leavitt BR. Epidemiology of Huntington disease. Handb Clin Neurol. 2017;144:31–46. https://doi.org/10.1016/B978-0-12-801893-4.00003-1.
Pfister EL, Kennington L, Straubhaar J, Wagh S, Liu W, DiFiglia M, et al. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington's disease patients. Curr Biol. 2009;19(9):774–8. https://doi.org/10.1016/j.cub.2009.03.030.
Kay C, Collins JA, Skotte NH, Southwell AL, Warby SC, Caron NS, et al. Huntingtin haplotypes provide prioritized target panels for allele-specific silencing in Huntington disease patients of European ancestry. Mol Ther. 2015;23(11):1759–71. https://doi.org/10.1038/mt.2015.128.
Castilhos RM, Augustin MC, Santos JA, Perandones C, Saraiva-Pereira ML, Jardim LB, et al. Genetic aspects of Huntington’s disease in Latin America. A systematic review. Clin Genet. 2016;89(3):295–303. https://doi.org/10.1111/cge.12641.
Baine FK, Kay C, Ketelaar ME, Collins JA, Semaka A, Doty CN, et al. Huntington disease in the South African population occurs on diverse and ethnically distinct genetic haplotypes. Eur J Hum Genet. 2013;21(10):1120–7. https://doi.org/10.1038/ejhg.2013.2.
•• Kay C, Collins JA, Caron NS, Agostinho LA, Findlay-Black H, Casal L, et al. A comprehensive haplotype-targeting strategy for Aallele-specific HTT suppression in Huntington disease. Am J Hum Genet. 2019;105(6):1112–25. https://doi.org/10.1016/j.ajhg.2019.10.011. Key considerations in any haplotype-dependent ASO therapeutic strategy.
Pulkes T, Papsing C, Wattanapokayakit S, Mahasirimongkol S. CAG-expansion haplotype analysis in a population with a low prevalence of Huntington’s disease. J Clin Neurol. 2014;10(1):32–6. https://doi.org/10.3988/jcn.2014.10.1.32.
Wild EJ, Tabrizi SJ. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017;16(10):837–47. https://doi.org/10.1016/S1474-4422(17)30280-6.
Iwamoto N, Butler DCD, Svrzikapa N, Mohapatra S, Zlatev I, Sah DWY, et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat Biotechnol. 2017;35(9):845–51. https://doi.org/10.1038/nbt.3948.
Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343(6256):364–6. https://doi.org/10.1038/343364a0.
Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006;47(7):3052–64. https://doi.org/10.1167/iovs.05-1443.
Talib M, Boon CJF. Retinal dystrophies and the road to treatment: clinical requirements and considerations. Asia Pac J Ophthalmol (Phila). 2020;9(3):159–79. https://doi.org/10.1097/APO.0000000000000290.
Murray SF, Jazayeri A, Matthes MT, Yasumura D, Yang H, Peralta R, et al. Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest Ophthalmol Vis Sci. 2015;56(11):6362–75. https://doi.org/10.1167/iovs.15-16400.
•• Chen KW, Chen JA. Functional roles of long non-coding RNAs in motor neuron development and disease. J Biomed Sci. 2020;27(1):38. https://doi.org/10.1186/s12929-020-00628-zKey concepts relevant to ASO therapeutics development across lncRNA-based neurological disorders.
Todd PK, Paulson HL. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol. 2010;67(3):291–300. https://doi.org/10.1002/ana.21948.
Thornton CA. Myotonic dystrophy. Neurol Clin. 2014;32(3):705–19, viii. https://doi.org/10.1016/j.ncl.2014.04.011.
Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta. 2015;1852(4):594–606. https://doi.org/10.1016/j.bbadis.2014.05.019.
•• Thornton CA, Wang E, Carrell EM. Myotonic dystrophy: approach to therapy. Curr Opin Genet Dev. 2017;44:135–40. https://doi.org/10.1016/j.gde.2017.03.007Key concepts relevant to ASO therapeutics design across lncRNA-based neurological disorders, particularly multisystem disorders and disorders where lncRNA pathology has wide-ranging primary and secondary impacts.
Musova Z, Mazanec R, Krepelova A, Ehler E, Vales J, Jaklova R, et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am J Med Genet A. 2009;149A(7):1365–74. https://doi.org/10.1002/ajmg.a.32987.
Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;69(2):385. https://doi.org/10.1016/0092-8674(92)90418-c.
Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488(7409):111–5. https://doi.org/10.1038/nature11362.
Pandey SK, Wheeler TM, Justice SL, Kim A, Younis HS, Gattis D, et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J Pharmacol Exp Ther. 2015;355(2):329–40. https://doi.org/10.1124/jpet.115.226969.
Mignon L. https://www.myotonic.org/sites/default/files/pages/files/Laurence-Mignon_IONIS-Update_2018-Conference.pdf. Myotonic Dystrophy Foundation meeting 2018.
Yadava RS, Yu Q, Mandal M, Rigo F, Bennett CF, Mahadevan MS. Systemic therapy in a RNA toxicity mouse model with an antisense oligonucleotide therapy targeting a non-CUG sequence within the DMPK 3'UTR RNA. Hum Mol Genet. 2020;29:1440–53. https://doi.org/10.1093/hmg/ddaa060.
Klein AF, Varela MA, Arandel L, Holland A, Naouar N, Arzumanov A, et al. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J Clin Invest. 2019;129(11):4739–44. https://doi.org/10.1172/JCI128205.
Christou M, Wengel J, Sokratous K, Kyriacou K, Nikolaou G, Phylactou LA, et al. Systemic evaluation of chimeric LNA/2'-O-methyl steric blockers for myotonic dystrophy type 1 therapy. Nucleic Acid Ther. 2020;30(2):80–93. https://doi.org/10.1089/nat.2019.0811.
Stepniak-Konieczna E, Konieczny P, Cywoniuk P, Dluzewska J, Sobczak K. AON-induced splice-switching and DMPK pre-mRNA degradation as potential therapeutic approaches for myotonic dystrophy type 1. Nucleic Acids Res. 2020;48(5):2531–43. https://doi.org/10.1093/nar/gkaa007.
Jamal GA, Weir AI, Hansen S, Ballantyne JP. Myotonic dystrophy. A reassessment by conventional and more recently introduced neurophysiological techniques. Brain. 1986;109(Pt 6):1279–96. https://doi.org/10.1093/brain/109.6.1279.
Labayru G, Diez I, Sepulcre J, Fernandez E, Zulaica M, Cortes JM, et al. Regional brain atrophy in gray and white matter is associated with cognitive impairment in Myotonic dystrophy type 1. Neuroimage Clin. 2019;24:102078. https://doi.org/10.1016/j.nicl.2019.102078.
Abramzon YA, Fratta P, Traynor BJ, Chia R. The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci. 2020;14:42. https://doi.org/10.3389/fnins.2020.00042.
Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–38. https://doi.org/10.1016/j.neuron.2013.07.033.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56. https://doi.org/10.1016/j.neuron.2011.09.011.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68. https://doi.org/10.1016/j.neuron.2011.09.010.
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–30. https://doi.org/10.1016/S1474-4422(12)70043-1.
Rademakers R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol. 2012;11(4):297–8. https://doi.org/10.1016/S1474-4422(12)70046-7.
Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;17(6):383–95. https://doi.org/10.1038/nrn.2016.38.
Cappella M, Ciotti C, Cohen-Tannoudji M, Biferi MG. Gene therapy for ALS-a perspective. Int J Mol Sci. 2019;20(18). https://doi.org/10.3390/ijms20184388.
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530–9. https://doi.org/10.1073/pnas.1318835110.
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90(3):535–50. https://doi.org/10.1016/j.neuron.2016.04.006.
Frazier KS. Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist’s perspective. Toxicol Pathol. 2015;43(1):78–89. https://doi.org/10.1177/0192623314551840.
Aupy P, Echevarria L, Relizani K, Zarrouki F, Haeberli A, Komisarski M, et al. Identifying and avoiding tcDNA-ASO sequence-specific toxicity for the development of DMD exon 51 skipping therapy. Mol Ther Nucleic Acids. 2020;19:371–83. https://doi.org/10.1016/j.omtn.2019.11.020.
Crooke ST, Baker BF, Witztum JL, Kwoh TJ, Pham NC, Salgado N, et al. The effects of 2'-O-Methoxyethyl containing antisense oligonucleotides on platelets in human clinical trials. Nucleic Acid Ther. 2017;27(3):121–9. https://doi.org/10.1089/nat.2016.0650.
Schoch KM, Miller TM. Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron. 2017;94(6):1056–70. https://doi.org/10.1016/j.neuron.2017.04.010.
Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington’s disease: what’s in the pipeline? Mov Disord. 2014;29(11):1434–45. https://doi.org/10.1002/mds.26007.
Ammala C, Drury WJ 3rd, Knerr L, Ahlstedt I, Stillemark-Billton P, Wennberg-Huldt C, et al. Targeted delivery of antisense oligonucleotides to pancreatic beta-cells. Sci Adv. 2018;4(10):eaat3386. https://doi.org/10.1126/sciadv.aat3386.
Huang Y. Preclinical and clinical advances of GalNAc-decorated nucleic acid therapeutics. Mol Ther Nucleic Acids. 2017;6:116–32. https://doi.org/10.1016/j.omtn.2016.12.003.
Tsimikas S, Viney NJ, Hughes SG, Singleton W, Graham MJ, Baker BF, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet. 2015;386(10002):1472–83. https://doi.org/10.1016/S0140-6736(15)61252-1.
Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A. 2013;110(6):2366–70. https://doi.org/10.1073/pnas.1221891110.
Michel L, Huguet-Lachon A, Gourdon G. Sense and antisense DMPK RNA foci accumulate in DM1 tissues during development. PLoS One. 2015;10(9):e0137620. https://doi.org/10.1371/journal.pone.0137620.
De Vivo DC, Bertini E, Swoboda KJ, Hwu WL, Crawford TO, Finkel RS, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842–56. https://doi.org/10.1016/j.nmd.2019.09.007.
• Testa CM, Jankovic J. Huntington disease: a quarter century of progress since the gene discovery. J Neurol Sci. 2019;396:52–68. https://doi.org/10.1016/j.jns.2018.09.022Includes framework for understanding challenges for early therapeutic intervention concepts, particularly for neurodegenerative disorders with a wide phenotype range.
• Aartsma-Rus A, Straub V, Hemmings R, Haas M, Schlosser-Weber G, Stoyanova-Beninska V, et al. Development of exon skipping therapies for Duchenne muscular dystrophy: a critical review and a perspective on the outstanding issues. Nucleic Acid Ther. 2017;27(5):251–9. https://doi.org/10.1089/nat.2017.0682Covers key ASO therapeutics challenges around treating the full patient population in a multi-etiology disorder, such as use of ASO cocktails within one therapeutic.
Rodrigues FB, Quinn L, Wild EJ. Huntington’s disease clinical trials corner: January 2019. J Huntingtons Dis. 2019;8(1):115–25. https://doi.org/10.3233/JHD-190001.
Bali T, Self W, Liu J, Siddique T, Wang LH, Bird TD, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatry. 2017;88(2):99–105. https://doi.org/10.1136/jnnp-2016-313521.
Hubers A, Just W, Rosenbohm A, Muller K, Marroquin N, Goebel I, et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol Aging. 2015;36(11):3117 e1-e6. https://doi.org/10.1016/j.neurobiolaging.2015.08.005.
Conte A, Lattante S, Zollino M, Marangi G, Luigetti M, Del Grande A, et al. P525L FUS mutation is consistently associated with a severe form of juvenile amyotrophic lateral sclerosis. Neuromuscul Disord. 2012;22(1):73–5. https://doi.org/10.1016/j.nmd.2011.08.003.
Deusterward R. https://patientworthy.com/2019/06/10/the-fda-congress-a-young-woman-dying-of-als-her-physician-and-her-parents-are-all-struggling-over-access-to-an-untested-therapy/. 2019.
Figueiredo M. https://alsnewstoday.com/2020/03/16/jacifusen-collaboration-funds-experimental-therapy-for-patients-with-rare-fus-als/. ALS News Today. 2020.
Thakur N. https://www.als.org/stories-news/deeper-look-als-association-efforts-speed-approval-gene-therapies. 2020.
• Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203–22. https://doi.org/10.1038/nrd.2016.246A look at a potential new ASO therapeutic class.
d'Ydewalle C, Ramos DM, Pyles NJ, Ng SY, Gorz M, Pilato CM, et al. The antisense transcript SMN-AS1 regulates SMN expression and is a novel therapeutic target for spinal muscular atrophy. Neuron. 2017;93(1):66–79. https://doi.org/10.1016/j.neuron.2016.11.033.
Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, et al. Stress granule assembly disrupts nucleocytoplasmic transport. Cell. 2018;173(4):958–71 e17. https://doi.org/10.1016/j.cell.2018.03.025.
Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367–71. https://doi.org/10.1038/nature22038.
Carroll JB, Bates GP, Steffan J, Saft C, Tabrizi SJ. Treating the whole body in Huntington’s disease. Lancet Neurol. 2015;14(11):1135–42. https://doi.org/10.1016/S1474-4422(15)00177-5.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neurology of Aging
Rights and permissions
About this article

Cite this article
Testa, C.M. Antisense Oligonucleotide Therapeutics for Neurodegenerative Disorders. Curr Geri Rep 11, 19–32 (2022). https://doi.org/10.1007/s13670-020-00341-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13670-020-00341-7