
Abstract
The mammalian (mechanistic) target of rapamycin (mTOR) is frequently activated in epithelial ovarian cancer and is regarded as an attractive therapeutic target. Preclinical investigations using mTOR complex 1 (mTORC1) inhibitors have demonstrated promising antitumor activity on ovarian cancer both in the setting of monotherapy and in combination with cytotoxic agents. Based on promising preclinical data, mTORC1 inhibitors are currently being evaluated in phase I/II trials in patients with ovarian cancer. In an effort to overcome resistance to mTORC1 inhibitors, the novel mTOR kinase inhibitors that inhibit both mTORC1 and mTORC2, or dual PI3K/mTOR inhibitor have recently been developed. In this report, we review the scientific rationale and evidence for the potential clinical benefits provided by mTOR inhibitors for patients with epithelial ovarian cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The mammalian (mechanistic) target of rapamycin (mTOR) is a serine/threonine kinase that plays a key role in cell growth and proliferation [1, 2]. In cells, mTOR acts as the catalytic subunit of two functionally distinct complexes, mTOR Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2). Because the mTOR signaling pathway is hyperactivated in a wide range of tumor types, including ovarian cancer, it is expected that inhibitors developed against mTOR may have broad therapeutic activity [1–3].
Rapamycin, a natural product isolated from the bacterium Streptomyces hygroscopicus, acts as an allosteric inhibitor of mTORC1. Through the specific inhibition of mTORC1 activity, rapamycin and its derivatives displays a multifunctional biologic activity profile, blocking cell proliferation, cell growth, cell survival, cell motility, cell differentiation, and angiogenesis [1–3].
On the basis of promising preclinical data, mTORC1 inhibitors are currently being evaluated in phase I/II trials in patients with ovarian cancer. This article highlights the scientific rationale for the use of mTORC1 inhibitors in ovarian cancer treatment and summarizes the available preclinical and clinical findings. Moreover, this review provides the current knowledge of the mechanism of resistance to mTORC1 inhibitors.
mTOR: Structure and Function
mTORC1 is composed of six proteins: mTOR, raptor, mLST8/GβL, PRAS40, and deptor. In response to extracellular signals, phosphatidylinositol-3-kinase (PI3K) phosphorylates PIP2 to generate PIP3, leading to recruitment of the AKT to the plasma membrane, where it is phosphorylated and activated. Activated AKT can directly activate mTORC1 by phosphorylation of mTOR at Ser2448, as well as cause indirect activation of mTORC1 by phosphorylating tuberous sclerosis complex 2 (TSC2), also called tuberin. Phosphorylation of TSC2 by AKT leads to the inhibition of the function of TSC1/TSC2 complex. When TSC1/TSC2 is active, TSC2 stimulates the conversion of Rheb (Ras homologue enriched in brain)-GTP to Rheb-GDP, which inactivates mTORC1. When TSC2 is phosphorylated and inactivated by AKT, Rheb-GTP stimulates the activity of mTORC1. Once activated, mTORC1 phosphorylates the translation-regulating factors S6K-1 (ribosomal S6 kinase-1) and 4EBP-1 (eukaryote translation initiation factor 4E binding protein-1). The activation of S6K-1 leads to the translation of mRNA encoding ribosomal proteins, elongation factors, and other proteins required for the transition from the G1 phase to the S phase of the cell cycle [1, 2]. The phosphorylation of 4EBP-1 also enhances translation of mRNA encoding cyclin D1, c-Myc, and hypoxia-inducible factor-1α (HIF-1α) mRNA, leading to cell cycle progression or angiogenesis [1–3].
The other mTOR complex, mTORC2, consists of six proteins: mTOR, rictor, mSIN1, protor, mLST8/GβL, and deptor [1]. The precise mechanism by which mTORC2 is activated remains unclear. However, a recent report suggests that PI3K is required for the activation of mTORC2 [4•]. Activated mTORC2 phosphorylates AGC kinases, such as AKT, serum and glucocorticoid-regulated kinase (SGK), and protein kinase C-α (PKCα), and controls growth by regulating lipogenesis, glucose metabolism, the actin cytoskeleton, and apoptosis [5].
mTORC1 Inhibitors
Rapamycin (sirolimus), a potent inhibitor of mTORC1, was isolated in 1975 from the bacterium Streptomyces hygroscopicus [6]. Rapamycin inhibits mTORC1 by first binding to the intracellular protein FK506 binding protein 12 (FKBP12). The resulting mTORC1 inhibitor-FKBP12 complex then binds to mTOR at the FKBP12-rapamycin-binding domain (FRB), and inhibits the serine/threonine kinase activity of mTORC1 by an allosteric mechanism. In contrast to mTORC1, mTORC2 does not bind to rapamycin/FKBP12 and thus is generally thought to be resistant to rapamycin treatment [1].
As rapamycin has very poor water solubility which limits its clinical use, several soluble ester analogs of rapamycin (rapalogs) were developed [2]. Currently, these analogs include temsirolimus, everolimus, and ridaforolimus (Fig. 1). Temsirolimus and everolimus are formulated for i.v. administration and oral administration, respectively. Ridaforolimus was initially developed as an i.v. formulation, but an oral formulation was subsequently developed [3, 7].

Chemical structures of rapamycin and its analogues
mTORC1 Signaling in Ovarian Cancer: Preclinical Findings
mTORC1 Activation in Epithelial Ovarian Cancer
According to previous reports, somatic mutations of PTEN, PIK3CA, PIK3R1, and AKT1 [8–12], amplification of AKT2 [13], and PTEN promoter hypermethylation [14] have been observed in ovarian cancer. Any one of these upstream genetic or epigenetic changes can lead to the increased activation of mTORC1 signaling in ovarian cancer.
Frequent mTORC1 activation in human epithelial ovarian cancer was first described by Altomare et al. in 2004. In their article, they reported that mTORC1 was activated in 55 % (17 of 31) of epithelial ovarian cancers [15]. Subsequently, using tissue microarrays of 98 primary ovarian cancers [52 clear cell carcinomas (CCC) and 46 serous adenocarcinomas (SAC)], Mabuchi et al. have reported that mTORC1 is more frequently activated in CCC than SAC (86.6 % vs. 50 %) [16]. These findings may be explained, at least in part, by the fact that a PIK3CA activating mutation occurred more frequently in CCC than in other histological subtypes of epithelial ovarian cancer. Activating mutations in PIK3CA have been described to occur in 33–40 % of OCCC and 12–20 % of endometrioid adenocarcinomas, which are significantly higher than that observed in SACs and mucinous adenocarcinomas [17].
Therapeutic Potential of mTORC1 Inhibitors in Ovarian Cancer
In the setting of monotherapy, treatment with mTORC1 inhibitors effectively attenuated the proliferation in human ovarian carcinoma cells in vitro and in vivo [18, 19]. Importantly, the growth-inhibitory effect of everolimus was striking in cells with high AKT/mTORC1 activity, but the effect was minimal in cells displaying low AKT/mTORC1 activity [18, 19]. These results indicate that ovarian cancer cells with elevated AKT/mTORC1 activity are more susceptible to mTORC1 inhibitors than cells displaying low AKT/mTORC1 activity. Thus, we believe that CCC or endometrioid adenocarcinomas which have frequent PI3K/AKT/mTOR activation might be most susceptible to PI3K/AKT/mTOR inhibition therapy [3].
In preclinical models of ovarian cancer, the mTORC1 inhibitors have been successfully combined with chemotherapeutic agents, including cisplatin [19], paclitaxel [20], carboplatin [21], or trabectedin [22]. The mTORC1 inhibitors have also shown promising activity in combination with other biologic agents including bevacizumab [23], MEK inhibitor [24], or EGFR inhibitor [25]. These data indicate that combining mTORC1 inhibition with cytotoxic agents or biologic agents could be a potentially effective approach in ovarian cancer treatment [3].
In xenograft models of ovarian cancer, treatment with mTORC1 inhibitors significantly attenuated tumor-angiogenesis by inhibiting the expression of HIF-1α and VEGF [18, 19]. Given the significant anti-proliferative and anti-angiogenic activities, mTORC1 inhibitors, especially orally bioavailable mTORC1 inhibitors, such as everolimus, may be a reasonable candidate as a maintenance therapy after front-line chemotherapy for preventing or delaying the development of recurrent disease [3].
mTORC1 Inhibitors in Ovarian Cancer: A Clinical Trial Review
On the basis of promising preclinical findings, temsirolimus, everolimus, and ridaforolimus are currently being tested in phase I/II clinical trials, either as single agents or in combination with cytotoxic or biological agents in patients with ovarian cancer (Table 1). Of these, the only study that has published result is the Gynecologic Oncology Group (GOG) phase II trial of temsirolimus (GOG 170-I). In this trial, temsirolimus monotherapy was evaluated in patients with persistent or recurrent epithelial ovarian and primary peritoneal malignancies [26•]. Of 60 enrolled patients, 54 were eligible for evaluation; 9.3 % experienced a partial response and 24.1 % had progression free survival (PFS) ≥6 months, which was just below that required to warrant inclusion in phase III studies in unselected patients [26•]. In this study, patients whose ovarian tumor has mTORC1 activity showed higher response rate compared to those without mTORC1 activity (PFS ≥6 months, 30.3 % vs. 11.8 %; response rate 11.8 % vs. 5.9 %). Therefore, the clinical activity of mTORC1 inhibitors should be evaluated further in patients with ovarian CCC, a chemoresistant histological subtype characterized by frequent hyperactivation of the mTORC1.
The GOG is currently conducting a phase II trial (protocol GOG0268) that incorporated temsirolimus in combination with carboplatin and paclitaxel followed by temsirolimus consolidation as a first-line therapy in patients with stage III-IV CCC of the ovary [27].
Common Toxicities Associated with mTORC1 Inhibitors
The mTORC1 inhibitors are generally well tolerated with most common side effects, including stomatitis, rash, fatigue, hyperglycemia, hyperlipidemia, hypercholesterolemia, and myelosuppression. Most of these side effects are mild and resolve with dose interruptions or dose reductions. Symptomatic noninfectious pneumonitis is a relatively uncommon class effect of mTOR inhibitors, which can be life-threatening. In a phase III clinical trial of temsirolimus in renal cell carcinoma patients, asthenia, rash, and anemia were the most common side effects with 51, 47, and 45 % of incidences, respectively [28]. In addition, a significant percentage of patients presented with metabolic disturbances, such as hyperlipidemia (27 %), hyperglycemia (26 %), and hypercholesterolemia (24 %). Respiratory symptoms, such as cough and dyspnea, were reported in approximately a quarter of the patients (26 and 28 % respectively), and stomatitis was observed in approximately 20 %.
Mechanism of Resistance to mTORC1 Inhibitors
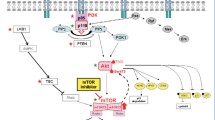
The precise mechanism responsible for the resistance to mTORC1 inhibitors remains unclear. However, two different pathways for mTORC1 inhibition-mediated AKT activation have been proposed as possible mechanisms (Fig. 2).

a Schematic representation of the mTOR signaling pathway. b The mechanisms of resistance to mTORC1 inhibitors
The first is mTORC2-independent insulin receptor substrate-1 (IRS1)-dependent AKT activation in response to mTORC1 inhibition (Fig. 2). mTORC1 phosphorylates and activates S6K-1, which in turn phosphorylates inhibitory sites (Ser636/639) on IRS-1, leading to the suppression of IRS-1 mediated activation of the PI3K/AKT pathway [3, 29]. The inhibition of mTORC1 by mTORC1 inhibitor results in the attenuation of negative feedback to IRS-1, leading to increased AKT activity (Fig. 2).
Another mechanism is mTORC2-mediated AKT activation in response to the treatment with mTORC1 inhibitors (Fig. 2). According to a recent preclinical study of ovarian CCC, the treatment with everolimus induces the activation of mTORC2, which leads to the activation of AKT [30•]. Moreover, the inhibition of mTORC2 activity by knocking down rictor attenuated everolimus-mediated AKT activation, enhanced the anti-tumor activity of everolimus, and prevented CCC cells from acquiring resistance to everolimus [30•]. These results indicate that mTORC2-mediated AKT activation is involved in the mechanism responsible for the acquired resistance to mTORC1 inhibitors. As mTORC2 is more frequently activated in CCCs than in SACs (71.2 % vs. 45.7 %, respectively), strategies aimed to inhibit the activity of mTORC2 have important roles as treatment for CCC [30•].
Recently, mTOR kinase inhibitors that can inhibit both mTORC1 and mTORC2 complexes have been developed [31]. Moreover, dual PI3K/mTOR inhibitors that can inhibit PI3K, mTORC1, and mTORC2 also have been developed [32]. Although these agents may have the potential to overcome resistance to mTORC1 inhibitors, they may be accompanied by unexpected, greater toxicities to normal tissues than mTORC1 inhibitors. However, in vivo treatment with these novel mTOR inhibitors displayed pronounced single-agent activity in multiple tumor models without causing apparent toxicities [31–33]. Some of these mTOR inhibitors are currently being examined in phase I/II clinical trials involving patients with solid malignancies, including ovarian cancer [34]. Future investigations may enable us to introduce these novel mTOR inhibitors for the clinical treatment of ovarian cancer.
Conclusions and Future Directions
mTORC1 is frequently activated in epithelial ovarian cancer, especially in CCC of the ovary. The mTORC1 inhibitors have shown promising activity in ovarian cancer cell lines and xenograft models, and the clinical activity of these agents either as single agents or in combination with cytotoxic or biologic agents is currently being evaluated in patients with ovarian cancer. Considering the mechanism of resistance to mTORC1 inhibitors, greater success also may be possible with novel molecules that target both mTORC1 and mTORC2. Given the potential toxicity of mTORC1-targeted therapy, it is now more important than ever to identify biomarkers that can be used to predict a patient’s sensitivity to mTORC1 inhibitors. Moreover, the identification of surrogate markers that are useful for monitoring the activity of mTORC1 inhibitors also is necessary. These challenges will aid the development of optimal, personalized mTORC1-tagreted therapies for ovarian cancer.
Abbreviations
- CCC:
-
Clear cell carcinoma
- SAC:
-
Serous adenocarcinoma
- mTOR:
-
mammalian target of rapamycin
- mTORC:
-
mTOR complex
- S6K-1:
-
Ribosomal S6 kinase-1
- 4E-BP1:
-
elF4E binding protein 1
- PI3K:
-
Phosphatidylinositol 3-kinase
- cisplatin:
-
cis-diamminedichloroplatinum
- PTEN:
-
Phosphatase and tensin homologue deleted on chromosome 10
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22.
Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6(9):729–34.
Mabuchi S, Hisamatsu T, Kimura T. Targeting mTOR signaling pathway in ovarian cancer. Curr Med Chem. 2011;18:2960–8.
Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144(5):757–68. This study is the first to show the mechanism by which mTORC2 is activated.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93.
Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo). 1975;28(10):727–32.
Mahalingam D, Sankhala K, Mita A, Giles FJ, Mita MM. Targeting the mTOR pathway using deforolimus in cancer therapy. Future Oncol. 2009;5(3):291–303.
Kolasa IK, Rembiszewska A, Janiec-Jankowska A, Dansonka-Mieszkowska A, Lewandowska AM, Konopka B, et al. PTEN mutation, expression and LOH at its locus in ovarian carcinomas. Relation to TP53, K-RAS and BRCA1 mutations. Gynecol Oncol. 2006;103(2):692–7.
Hashiguchi Y, Tsuda H, Inoue T, Berkowitz RS, Mok SC. PTEN expression in clear cell adenocarcinoma of the ovary. Gynecol Oncol. 2006;101(1):71–5.
Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;11(8):2875–8.
Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61(20):7426–9.
Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–44.
Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, et al. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci U S A. 1992;89(19):9267–71.
Yang HJ, Liu VW, Wang Y, Tsang PC, Ngan HY. Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC Cancer. 2006;6:212.
Altomare DA, Wang HQ, Skele KL, De Rienzo A, Klein-Szanto AJ, Godwin AK, et al. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene. 2004;23(34):5853–7.
Mabuchi S, Kawase C, Altomare DA, Morishige K, Sawada K, Hayashi M, et al. mTOR is a promising therapeutic target both in cisplatin-sensitive and cisplatin-resistant clear cell carcinoma of the ovary. Clin Cancer Res. 2009;15(17):5404–13.
Kuo KT, Mao TL, Jones S, Veras E, Ayhan A, Wang TL, et al. Shih IeM. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009;174(5):1597–601.
Mabuchi S, Altomare DA, Connolly DC, Klein-Szanto A, Litwin S, Hoelzle MK, et al. RAD001 (Everolimus) delays tumor onset and progression in a transgenic mouse model of ovarian cancer. Cancer Res. 2007;67(6):2408–13.
Mabuchi S, Altomare DA, Cheung M, Zhang L, Poulikakos PI, Hensley HH, et al. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin Cancer Res. 2007;13(14):4261–70.
Jiang H, Feng Y. Hypoxia-inducible factor 1alpha (HIF-1alpha) correlated with tumor growth and apoptosis in ovarian cancer. Int J Gynecol Cancer. 2006;16 Suppl 1:405–12.
Schlosshauer PW, Li W, Lin KT, Chan JL, Wang LH. Rapamycin by itself and additively in combination with carboplatin inhibits the growth of ovarian cancer cells. Gynecol Oncol. 2009;114(3):516–22.
Mabuchi S, Hisamatsu T, Kawase C, Hayashi M, Sawada K, Mimura K, et al. The activity of trabectedin as a single agent or in combination with everolimus for clear cell carcinoma of the ovary. Clin Cancer Res. 2011;17(13):4462–73.
Huynh H, Teo CC, Soo KC. Bevacizumab and rapamycin inhibit tumor growth in peritoneal model of human ovarian cancer. Mol Cancer Ther. 2007;6(11):2959–66.
Kinross KM, Brown DV, Kleinschmidt M, Jackson S, Christensen J, Cullinane C, et al. In vivo activity of combined PI3K/mTOR and MEK inhibition in a Kras(G12D);Pten deletion mouse model of ovarian cancer. Mol Cancer Ther. 2011;10(8):1440–9.
Glaysher S, Bolton LM, Johnson P, Atkey N, Dyson M, Torrance C, et al. Targeting EGFR and PI3K pathways in ovarian cancer. Br J Cancer. 2013;109(7):1786–94.
Behbakht K, Sill MW, Darcy KM, Rubin SC, Mannel RS, Waggoner S, et al. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: a Gynecologic Oncology Group study. Gynecol Oncol. 2011;123(1):19–26. The only published result of phase II study of mTORC1 inhibitor.
NCT01196429. Clinical Trials.gov. A Service of the U.S. National Institute of Health. http://www.clinicaltrials.gov (Accessed 31 Mar 2014).
Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356(22):2271–81.
Takano A, Usui I, Haruta T, Kawahara J, Uno T, Iwata M, et al. Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol Cell Biol. 2001;21(15):5050–62.
Hisamatsu T, Mabuchi S, Matsumoto Y, Kawano M, Sasano T, Takahashi R, et al. Potential role of mTORC2 as a therapeutic target in clear cell carcinoma of the ovary. Mol Cancer Ther. 2013;12(7):1367–77. This study is the first to investigate the role of mTORC2 in the acquired resistance to mTORC1 inhibitor in ovarian cancer.
Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene. 2010;29(26):3733–44.
Ghadimi MP, Lopez G, Torres KE, Belousov R, Young ED, Liu J, et al. Targeting the PI3K/mTOR axis, alone and in combination with autophagy blockade, for the treatment of malignant peripheral nerve sheath tumors. Mol Cancer Ther. 2012;11(8):1758–69.
Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70(1):288–98.
NCT01936363. Clinical Trials.gov. A Service of the U.S. National Institute of Health. http://www.clinicaltrials.gov (Accessed 31 Mar 2014).
Acknowledgments
The authors thank Yuko Nishimura for providing secretarial assistance.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Seiji Mabuchi, Tomoyuki Sasano, Mahiru Kawano, Hiromasa Kuroda, and Tadashi Kimura declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Gynecological Cancer
Rights and permissions
About this article

Cite this article
Mabuchi, S., Sasano, T., Kawano, M. et al. Targeting mTOR Signaling in Ovarian Cancer. Curr Obstet Gynecol Rep 4, 11–17 (2015). https://doi.org/10.1007/s13669-014-0102-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13669-014-0102-y