
Abstract
Purpose of Review
Interstitial lung diseases are a heterogeneous group of disorders characterized by varying degrees of inflammation and scarring of the lung parenchyma. Diagnosis can be challenging and requires careful multidisciplinary appraisal of carefully obtained history, physical examination, serological profile, imaging, and, at times, lung tissue. We aim to provide a roadmap for the diagnosis of ILD.
Recent Findings
The diagnostic criteria for IPF, which is the deadliest form of idiopathic interstitial pneumonia, and HRCT pattern classification have been updated. Transbronchial cryobiopsies are becoming more prevalent but overall diagnostic yield compared to surgical lung biopsy is not known.
Summary
A technically adequate high-resolution CAT scan of the chest (HRCT) is a central element but a multidisciplinary evaluation of all available evidence is fundamental for the diagnosis of ILD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Interstitial lung disease (ILD) (or diffuse parenchymal lung disease) is a heterogeneous group of non-neoplastic disorders characterized by damage to the lung parenchyma with inflammation and fibrosis leading to progressive loss of function and death. They share a common clinical presentation that includes persistent cough and dyspnea on exertion or at rest associated with physiological impairment measured by pulmonary function tests, hypoxemia and characteristic radiographic and histological patterns [1]. An epidemiological study from Bernalillo County in New Mexico estimated that the overall incidence of ILD is approximately 26.1 and 31.5 cases/100,000 in females and males, respectively [2, 3]. Idiopathic Pulmonary Fibrosis (IPF) alone is estimated to have a prevalence of approximately 495 cases/100,000-person years in 2011 [3].
Despite the many similarities, each of the more than 150 disorders grouped under this umbrella has very distinct etiologies and natural history (Fig. 1). Treatment and various management decisions are often diagnosis-specific and may vary considerably [4]. Distinguishing the specific ILD diagnosis requires a very systematic process that integrates clinical data, imaging, and sometimes, histology [5].

Interstitial lung disease classification
In this manuscript, we suggest a general roadmap for the diagnostic approach of a patient presenting with ILD. We will highlight the important clues provided by a detailed history, physical examination as well as the initial physiological and serological assessments. We will also emphasize the importance of appropriate imaging techniques and discuss integration of high-resolution CT scan (HRCT) patterns in the ILD diagnostic algorithm and, lastly, we will discuss the role of lung biopsy and the importance of a multidisciplinary approach.
History and Physical Examination
Demographics and Family History
IPF is the most common of the idiopathic interstitial pneumonias and is thought to be a disease related to aging [6, 7]. Sporadic cases of IPF occur predominantly among Caucasians older than age 50 and it is slightly more prevalent among males [7]. Approximately 5 to 20% of IPF cases are “familial” [8], with two or more members of the same family being affected. The clinical characteristics of sporadic and familial cases of IPF are largely the same [9]. In contrast, ILD related to connective tissue disorders (CTD-ILD) are typically more prevalent among younger adults and racial minorities, and a major risk factor for developing a CTD is having a first degree relative with the diagnosis [10]. Sarcoidosis has higher prevalence among African American women and its incidence also clusters in families [11]. Lymphangioleyomyomatosis (LAM) occurs exclusively in premenopausal women [12]. Hermasnky-Pudlak Syndrome is a rare genetic condition that has a higher incidence among Puerto Ricans [13] and, although it has a clinical presentation that is very similar to IPF, it typically affects much younger individuals [14].
Exposures
A detailed history with type, duration, and intensity of exposure is a crucial step in the evaluation of a patient with ILD. Certain conditions such as acute hypersensitivity pneumonitis (AHP) and acute silicosis are associated with short-term high-intensity exposure while others, such as asbestosis, smoking-related-ILDs, and chronic hypersensitivity pneumonitis (CHP) are usually the result of many years of exposure. CHP is often related to exposure to an obvious antigen such as avian proteins but in some cases, the cause can be elusive (Table 1) [15]. The importance of identifying the inciting antigen needs to be underscored as failure to do so may be an independent predictor of worse prognosis among patients with CHP [16].
Respiratory bronchiolitis-ILD (RB-ILD) and desquamative interstitial pneumonia (DIP) are virtually always related to cigarette smoking. Similarly, pulmonary Langerhans cell granulomatosis (PLCH) is typically a disease of younger smokers that present with dyspnea on exertion and nodular and cystic lung disease in HRCT.
Pneumoconiosis is a group of chronic lung diseases caused by occupational exposure to different dusts/fibers such as silica, coal, or asbestos. Silicosis can present in a spectrum ranging from acute silicosis, accelerated silicosis and more chronic forms depending upon the type and duration of exposure [17]. In the case of interstitial lung disease related to asbestos exposure (asbestosis), the latency period for disease development is inversely proportional to the degree of exposure and usually ranges from 10 to 20 years [18]. Similarly, the progression of coal worker’s pneumoconiosis (CWP) into massive fibrosis is influenced by duration and total cumulative exposure as well as concomitant infections [19].
Hundreds of drugs have been listed as potential causes of interstitial lung disease and toxicity can be idiosyncratic or dose-dependent mediated as either a direct effect or of particular metabolite (s). Unfortunately, there are no specific clinical, radiological or even histological features for the diagnosis. Patients may present with respiratory failure as in sulfasalazine-induced acute pneumonitis or a more chronic fibrosing lung disease as in amiodarone or nitrofurantoin toxicity. Hence, when faced with a case of ILD, one must take a careful and detailed history of drug intake that includes dosing, timing, and latency. Given the ever-evolving number of agents, particularly biologicals used for the treatment of malignancies and auto-immune disorders, it is challenging for clinicians to stay updated on emerging reports of lung toxicity. Resources such as pneumotox.com curate a comprehensive and frequently updated review of the literature on drug-related lung toxicity.
Physical Examination
A careful physical examination may offer clues to the specific diagnosis of a patient presenting with interstitial lung disease. Lung auscultation often demonstrates bibasilar crackles that sound like two pieces of velcro™ being slowly pulled apart in conditions such as IPF, fibrotic idiopathic NSIP, scleroderma, rheumatoid arthritis, and asbestosis. The presence of wheezing suggests some overlap with diseases that affect the airways such as combined pulmonary fibrosis and emphysema (CPFE), eosinophilic granulomatosis with polyangiitis, or concomitant asthma. Wheezing may also be present in cases of bronchiolitis and CHP in addition to “squeaks” and “pops” on inspiration [20, 21]. Digital clubbing is a non-specific finding that may be present in up to 50% of IPF patients at the time of diagnosis [5]. Most connective tissue disorders and other systemic conditions may be associated with subtle physical findings that are clues to the diagnosis (Table 2). As an example, systemic sclerosis is often associated with facial telangiectasias, reduced mouth aperture, skin thickening, and digital ulcerations. Inflammatory myopathies may be associated with mechanic’s hands and dermatomyositis may present with an erythematous rash over the knuckles (Gottron’s papules).
Pulmonary Function Tests
Evaluation of the lung physiology is of limited diagnostic value if not integrated in the overall clinical context. Pulmonary function testing is more often used to assess disease severity, progression, and response to therapy. Most cases of ILD present with a reduction in static lung volumes. The spirogram usually demonstrates a proportional reduction of forced vital capacity (FVC) and forced expiratory volume in the first second (FEV1) that characterizes restriction. Obstructive abnormalities with/without elevation of lung volumes are less frequent but may help narrow the diagnosis to one of the conditions that may affect the airways (Table 3) [22,23,24]. Most patients with IPF have a substantial smoking history, so the presence of concomitant emphysema is not infrequent. Patients with combined pulmonary fibrosis and emphysema (CPFE) may present with relatively preserved lung volumes, which may delay the recognition of the underlying pathology [25, 26]. Often, the only clue is a disproportionally low diffusion capacity for carbon monoxide (DLCO), which can also be seen in ILD patients with secondary pulmonary arterial hypertension (PAH).
Oxygen levels should be measured at rest and during exertion during the clinical investigation of patients with ILD [5]. The finding of hypoxemia during exertion is a common finding and may portent worse prognosis for patients with ILD [27].
Serological Evaluation
Data from a series of registries suggest that connective tissue diseases are the third most common cause of interstitial lung disease (CTD-ILD) [28]. In the USA, it is estimated that the prevalence of CTD-ILD is approximately 7.1 and 11.6/100,000 persons for men and women, respectively [2] and a significant number of patients with rheumatoid arthritis and scleroderma will have evidence of interstitial abnormalities in chest imaging during the course of their disease [29, 30]. Furthermore, ILD is a leading cause of death among patients with systemic sclerosis and rheumatoid arthritis [31, 32]. The determination of whether a patient presenting with ILD has a CTD can be very challenging. An ILD may be the sole initial manifestation of a CTD [33, 34] and the absence of extrapulmonary signs and symptoms may delay clinical suspicion and subsequent investigation. Hence, serological evaluation is strongly recommended for all patients presenting with an ILD despite the lack of high-quality evidence to inform how comprehensive that evaluation ought to be for a patient without extrapulmonary manifestations. The ATS/ERS/JRS/ALAT guidelines on the diagnosis of IPF issues a broad recommendation for serological testing, with the majority of the panelists listing C-reactive protein, erythrocyte sedimentation rate, ANA (by immunofluorescence), RF, myositis panel, and anti-CCP as part of the initial panel [35••]. Bahmer and colleagues propose a stepwise approach whereby all ILD patients should have an ANA (tested by indirect immunofluorescence) along with Ro/SSA, La/SSB, and Jo-1 as these autoantibodies may be missed by the ANA screening. Finally, they argue that the elevated prevalence of rheumatoid arthritis and the fact that an interstitial lung disease may be the initial presentation of ANCA-mediated vasculitis, justifies that one also obtains CCP, RF, and ANCAs as part of the initial evaluation [36]. Further testing including the t-RNA synthetase-related autoantibodies should be checked if the suspicion is corroborated by physical examination findings and/or elevation of muscle enzymes. Positive serologies without overt clinical manifestations of connective tissue disease in the setting of ILD may necessitate a rheumatological evaluation. We have a low threshold for Rheumatology referral for younger patients, particularly those belonging to a racial minority or those with HRCT findings that are atypical for UIP. A Rheumatologist may unveil unrecognized, and sometimes subtle, physical findings such as nail fold capillary abnormalities, diminished mouth aperture, or mechanic’s hands.
Imaging
Although a plain chest radiograph may demonstrate the presence of ILD, its value is usually limited to suggesting whether the disease affects predominantly the upper or lower zones. High-resolution computer assisted tomography (HRCT) has become the most important element in the diagnostic algorithm of interstitial lung diseases and a technically adequate study can be diagnostic, abrogating the need for a lung biopsy [37].
HRCT images should be obtained with the patient in the prone position, with thin sections (< 2 mm) using a high spatial resolution reconstruction algorithm in both full inspiration and expiration [38, 39]. Volumetric CT acquisition is preferred to non-contiguous imaging since it improves characterization and delineates extent and distribution of the disease [40]. Images obtained in full inspiration decrease lung attenuation and help with better visualization interstitial abnormalities, particularly reticular and ground glass opacities [39]. On the other hand, expiratory views may identify mosaic attenuation and air trapping that can be associated with chronic hypersensitivity pneumonitis or certain connective tissue diseases [41]. This is a very important element, as CHP is a common mimicker of IPF [42].
Prone positioning helps in reducing dependent atelectasis, better visualization of the posterior lung bases, and recognition of findings such as fine reticulations which can be missed with supine imaging. Prone images may also facilitate confirming the diagnosis of honeycombing by reducing interobserver variability [43, 44].
The main HRCT patterns seen in ILD include honeycombing, reticular opacities, traction bronchiectasis, and ground glass opacities. A given pattern may be related to a specific diagnosis (Table 4), and hence requires familiarity with terminology and features. Honeycombing is defined as clustered, thick-walled cystic spaces of similar diameters, generally measuring between 3 and 5 mm, but occasionally up to 25 mm in size [45]. A single subpleural layer of two or three contiguous cysts is sufficient for diagnosing honeycombing [46]. Honeycombing noted on CT corresponds histologically to honeycomb cysts from revised alveolar spaces and bronchiolectasis but microscopic honeycombing can be present in the absence of visible honeycombing in a HRCT.
Traction bronchiectasis represents dilation of the bronchioles secondary to surrounding retractile fibrosis [45] and may be associated with worse outcomes [47]. It can be challenging to differentiate peripherally-located traction bronchiectasis from honeycombing, so it is recommended that sequential multiplanar images enhanced by post processing algorithms are used when this question arises [48••].
Reticular opacities can be either fine or coarse interlaced curvilinear lines that are associated with thickening of the pulmonary interstitium and, at times, microscopic honeycombing.
Ground glass opacities are areas of obfuscation of the lung parenchyma by faint white opacities through which underlying bronchial and vascular structures are still identifiable. It may represent alveolar filling processes such as what is observed with desquamative interstitial pneumonia (DIP) or interstitial inflammation as seen in NSIP (idiopathic or secondary to CTD).
A white paper from the Fleischner society on diagnostic criteria for IPF [48••] recommends that HRCT patterns be classified based on whether it is consistent with UIP or otherwise (Table 5). Features such as peribronchovascular opacities and cystic changes are suggestive of “diagnosis other than IPF” such as sarcoidosis and LAM or PLCH, respectively.
Appraisal of the HRCT pattern and the predominant location of the ILD (Table 6) is a critical step, but it is ultimately up to the clinician and often to the multidisciplinary team to arrive at the most likely diagnosis by integrating all the clinical and imaging data available.
Bronchoalveolar Lavage, Transbronchial Biopsy, Bronchoscopic Cryobiopsy, and Surgical Lung Biopsy
Occasionally, a diagnosis cannot be defined even after careful review of the clinical history, physical examination, imaging, and serological data. As one ponders on the need for an invasive procedure, the main considerations are diagnostic yield, safety, and whether the results are likely to affect the management.
Bronchoalveolar lavage (BAL) is considered to be safe in patients with ILD [49] but there is data suggesting an increased risk of acute exacerbations following the procedure in patients with IPF [50]. When performed, no less than 100 mL and no more than 300 mL of normal saline should be instilled in a subsegment [51•]. In general, BAL findings are considered to be neither sensitive nor specific in ILD but in certain conditions, it may provide enough evidence to support (or refute) a diagnosis. A milky appearing fluid with PAS-positive debris is consistent with pulmonary alveolar proteinosis and fluid aliquots that are increasingly bloody-appearing are seen in patients with diffuse alveolar hemorrhage. Sarcoidosis has a higher proportion of lymphocytes and stable IPF patients tend to have a higher neutrophil count [52]. A differential eosinophil count ≥ 25% suggests eosinophilic pneumonia, whereas 5% or more of CD1a positive cells corroborates the diagnosis of Pulmonary Langerhans’ cell histiocytosis [53]. The recent updated guidelines on the diagnosis of IPF suggest that a BAL be considered for patients with ILD of unknown cause presenting with a HRCT pattern different than UIP [35••].
The utility of a transbronchial biopsy (TBBx) in the diagnosis of ILD is hampered by the small size of specimens and frequent presence of crushing artifacts, which can be minimized by use of an alligator forceps, although a cup forceps may be more likely to obtain alveolated tissue [54]. In a recent series, more than half of the specimens obtained via TBBx were deemed inadequate [55]. Overall, TBBx is felt to be safe, and recent report estimates the risk of pneumothorax to be approximately 7% in patients with pulmonary fibrosis and it was directly associated with disease severity [56]. TBBx has relatively low diagnostic yield in ILD but when combined with clinical and HRCT data and in the context of a multidisciplinary discussion among experts, it may provide enough evidence for a confident diagnosis in 20–30% of cases [55]. Genomic analysis and machine learning technology may improve the utility of TBBx for the diagnosis of UIP but this promising methodology has not been broadly validated [57].
Bronchoscopic cryobiopsies require deep sedation and endotracheal rigid intubation in the operating room. Biopsies are obtained via a flexible cryoprobe that freezes the tissue for samples that are 0.5 to 1 cm in size [58]. The advantages of the CryoBx over TBBx are related to the larger sizes of the specimens which allow for a better evaluation of the overall pattern of lesion and lack of crushing artifact [59]. The overall diagnostic yield of CryoBx is unknown as there has been no study comparing it to the more invasive surgical lung biopsy (SLBx). However, the diagnostic yield of CryoBx in isolation and in the context of a multidisciplinary discussion is quite good and estimated to be approximately 83% and 79% respectively. Safety concerns remain an issue, with incidence of pneumothorax and moderate to severe bleeding reported to be 12% and 39% respectively [60•, 61].
A surgical lung biopsy may be indicated in cases where the diagnosis cannot be confidently established with data from a careful clinical evaluation, radiology, and/or less invasive histological sampling. The 2011 ATS/ERS/JRS/ALAT Guidelines on the diagnosis and management of IPF suggests that a surgical lung biopsy be performed in cases where the HRCT demonstrates a pattern different than “definite UIP” and in the absence of other data suggesting an alternative diagnosis [5, 62]. The updated 2018 guidelines maintained that recommendation [35••]. One can also consider a SLBx in a patient with established diagnosis of ILD presenting with new and atypical features [62]. The mortality associated with a SLBx can be substantial. Acutely ill patients undergoing the procedure may have in-hospital mortality rates in excess of 15%. The risk of death related to an elective SLBx in clinically stable patients is estimated to be less than 5% but this number varies greatly in the literature and is likely related to patient selection and experience of the surgical team [63, 64]. The risk of poor outcomes following a SLBx is higher among males and increases with age and the number of comorbidities [63]. Most SLBx are now been performed thoracoscopically and at least two lobes should be sampled given the frequent histologic variability noted in cases of ILD [65]. The findings of a surgical biopsy should not be considered the final diagnosis in a patient with ILD as many diseases may share histological patterns (Table 7). The biopsy samples should always be reviewed in the context of a multidisciplinary appraisal of all the available clinical and imaging data [5, 66]. Unfortunately, in many instances, only a provisional diagnosis can be issued and prospective clinical surveillance for further clues will remain an important aspect of patient management.
Conclusions and Summary of the Diagnostic Algorithm
Invariably, it is the primary care provider that suspects the presence of lung disease. We recommend that any patient presenting with a constellation of manifestations that includes dyspnea on exertion/exercise limitation, persistent cough, digital clubbing, and/or crackles on lung auscultation be investigated for ILD. Once ILD is suspected, the first step is a comprehensive history and physical examination, followed by pulmonary function tests, serologies, and a HRCT. In most cases, a diagnosis can be made with a good level of confidence; otherwise, if considering an invasive procedure, one ought to conduct a multidisciplinary discussion, which should be repeated if tissue is actually obtained (Fig. 2). It is important to underscore the fact that not all MDDs end in a consensus diagnosis. Obtaining a biopsy may always not be feasible based on age, comorbidities, severity of the disease, or patient preferences. In such cases, one may have to establish a “provisional” diagnosis based on the available evidence and continue to seek for clues that may come forth over time. Important clues include response to empirical therapies and the development of systemic manifestations of underlying connective tissue diseases.

Diagnostic algorithm. PFTs, pulmonary function testing; BAL, bronchoalveolar lavage; TBBx, transbronchial biopsy; TBCryoBx, transbronchial cryobiopsy
Integrating the Data—Case Examples
Case 1
History and Physical Examination
A 70-year-old Caucasian female presented for a second opinion on a diagnosis of IPF that had been established via surgical lung biopsy in another institution. She had a 9-month history of dyspnea on exertion in addition to mild persistent asthma, fibromyalgia, and irritable bowel syndrome. There was no family history of ILD or CTD. She worked as a waitress and had daily exposure to second-hand smoking. She owned a plum-headed parakeet for 10 years. On physical examination, there were bibasilar inspiratory crackles and a few inspiratory and expiratory squeaks in the upper zones.
Pulmonary Function Testing
FVC 73%, FEV1 78%, ratio 0.81, TLC 70%, DLCO 55%. No hypoxemia in a 6-min walk test.
Serological Evaluation
ANA, CCP, RF, Scl-70, SSA, SSB, RNP, c-ANCA, p-ANCA, Jo1, MI 2, PL7, PL12, EJ, OJ, SRP, KU, U2 SN RNP: negative
HRCT
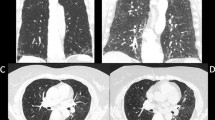
Interstitial changes more pronounced in the lower lung zones, there was no overt honeycombing and the predominant finding is ground glass opacities. Expiratory views demonstrated areas of lobular air trapping (Fig. 3).

HRCT images in inspiration (3.A) and expiration (3.B). Note diffuse patchy ground glass opacities in inspiration and mosaic attenuation in expiration, consistent with air trapping (white arrows)
Lung Biopsy
Findings included areas of normal lung interspersed with areas affected by dense fibrosis and remodeling. A few fibroblast foci and airway-centered poorly formed non-caseating granulomas were present (Fig. 4).

High power view of a poorly formed non-caseating granuloma (black arrow)
Multidisciplinary Discussion
The patient’s age is certainly consistent with original diagnosis of IPF. She had significant exposures and smoking-related ILD such as RB-ILD and DIP may occur with second-hand exposure to cigarette smoking. Her exposure to a pet bird and the presence of upper zone bronchiolar sounds (squeaks) are concerning for CHP. She had no manifestations of a CTD and a negative serological panel makes this diagnosis unlikely. Her PFTs demonstrate a restrictive pattern that is consistent with the presence of ILD. The lack of airflow obstruction does not rule out the possibility of CHP or concomitant emphysema or asthma. Her HRCT findings are very important. She has extensive areas of ground glass opacities but also some fibrotic changes manifested as reticulation and mild traction bronchiectasis. The absence of honeycombing is also relevant as is the presence of mosaic attenuation and areas of clear lobular air trapping. In the most recent classification proposed by the Fleischner Society [48••], her HRCT would be classified as “most consistent with a non-IPF diagnosis”. Her long history of exposure to a pet bird, the physical examination findings, and the presence of air trapping in the HRCT does support a diagnosis of CHP and the findings in surgical lung biopsy, although consistent with CHP, did not really add any significant information to what was already known without it. This case is an example of how careful appraisal of all the evidence arising from a comprehensive exposure history and a technically adequate HRCT may abrogate the need for an invasive procedure.
Final Diagnosis
Chronic hypersensitivity pneumonitis
Case 2
History and Physical Examination
Sixty-one-year-old Caucasian male presented with 18-month history of dyspnea and non-productive cough. No occupational or household exposures. No family history of ILD or CTD. Never smoker. Lung auscultation revealed bibasilar inspiratory crackles.
Pulmonary Function Testing
FVC 65%, FEV1 71%, ratio .77, TLC 68%, DLCO 59%. Six-minute walk test: nadir SpO2 was 87%. Maximum HR 110 bpm
Serological Evaluation
ANA, CCP, RF, Scl-70, SSA, SSB, RNP, c-ANCA, p-ANCA, Jo1, MI 2, PL7, PL12, EJ, OJ, SRP, KU, U2 SN RNP: negative
HRCT
Extensive subpleural reticulation that predominates in the lower lung zones and areas of honeycombing. No air trapping on expiratory views. Findings consistent with “typical UIP pattern” (Fig. 5).

Predominantly lower lobe and subpleural reticulation (white arrows), traction bronchiectasis (arrow heads) and areas of honeycombing (black arrows). Findings are consistent with “typical UIP pattern”
Multidisciplinary Discussion
This is an older Caucasian male without any significant exposures and no evidence of an autoimmune disease presenting with dyspnea on exertion, cough, restrictive PFTs, hypoxemia on exertion, and UIP pattern in a good quality HRCT. In this case, there is no need for further testing and a lung biopsy is not indicated.
Final Diagnosis
Idiopathic pulmonary fibrosis
Case 3
History and Physical Examination
Thirty-six-year-old African American female presented with 2 years of progressive dyspnea. She endorsed frequent episodes of heartburn and the sensation that her food “is not going down”. Her fingers turn blue and hurt during the colder months. An aunt died of “lupus”. She denied any household or occupational exposures. The physical examination revealed thickening of the skin on her face with limited mouth aperture. She had active Raynaud’s and skin changes consistent with sclerodactyly. Lung auscultation demonstrated bibasilar inspiratory crackles. Heart auscultation demonstrated a loud S2.
Pulmonary Function Testing
FVC 48%, FEV1 53%, TLC 60%, DLCO 30%. Six-minute walk test: nadir SpO2 was 82%.
Serological Evaluation
Positive ANA (1:1280, nucleolar pattern) and Scl-70. All others: negative
HRCT
Extensive faint ground glass opacities predominating in the lower lung zones, along with few areas of coarse reticulation and no overt honeycombing. Esophagus appears patulous (see Fig. 6).

Extensive faint ground glass opacities (white arrow) predominating in the lower lung zones, along with few areas of coarse reticulation (black arrows) and no overt honeycombing. The esophagus is patulous (arrow head)
Multidisciplinary Discussion
This is a young African American female with a family history of CTD presenting with manifestations suspicious for Systemic Sclerosis, which is corroborated by serological results. The findings of a loud S2 on physical examination and disproportionally low DLCO are concerning for secondary pulmonary arterial hypertension, which has a high incidence in Systemic Sclerosis. Her HRCT demonstrates a pattern consistent with fibrotic NSIP, which can be seen in cases of ILD secondary to CTD. A patulous esophagus also raises the suspicion for systemic sclerosis. Taken together, the data is consistent with ILD secondary to systemic sclerosis, complicated by esophageal dysmotility and probable secondary pulmonary arterial hypertension. In this case, a biopsy is not indicated as it would neither change the diagnosis nor affect management.
Final Diagnosis
ILD secondary to underlying CTD (systemic sclerosis)
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150(4):967–72.
Raghu G, Chen SY, Yeh WS, Maroni B, Li Q, Lee YC, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2(7):566–72.
Meyer KC. Diagnosis and management of interstitial lung disease. Translational Respiratory Medicine. 2014;2:4.
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–61.
Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810–6.
Lawson WE, Loyd JE. The genetic approach in pulmonary fibrosis: can it provide clues to this complex disease? Proc Am Thorac Soc. 2006;3(4):345–9.
Lee HL, Ryu JH, Wittmer MH, Hartman TE, Lymp JF, Tazelaar HD, et al. Familial idiopathic pulmonary fibrosis: clinical features and outcome. Chest. 2005;127(6):2034–41.
Spagnolo P, Cordier JF, Cottin V. Connective tissue diseases, multimorbidity and the ageing lung. Eur Respir J. 2016;47(5):1535–58.
Baughman RP, Field S, Costabel U, Crystal RG, Culver DA, Drent M, et al. Sarcoidosis in America. Analysis based on health care use. Annals ATS. 2016;13(8):1244–52.
Sullivan EJ. Lymphangioleiomyomatosis: a review. Chest. 1998;114(6):1689–703.
Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome). N Engl J Med. 1998;338(18):1258–64.
Vicary GW, Vergne Y, Santiago-Cornier A, Young LR, Roman J. Pulmonary fibrosis in Hermansky-Pudlak syndrome. Annals ATS. 2016;13(10):1839–46.
Patel AM, Ryu JH, Reed CE. Hypersensitivity pneumonitis: current concepts and future questions. J Allergy Clin Immunol. 2001;108(5):661–70.
Fernandez Perez ER, Swigris JJ, Forssen AV, Tourin O, Solomon JJ, Huie TJ, et al. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest. 2013;144(5):1644–51.
Leung CC, Yu IT, Chen W. Silicosis. Lancet. 2012;379(9830):2008–18.
Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1666–80.
Castranova V, Vallyathan V. Silicosis and coal workers’ pneumoconiosis. Environ Health Perspect. 2000;108(Suppl 4):675–84.
Sellares J, Hernandez-Gonzalez F, Lucena CM, Paradela M, Brito-Zeron P, Prieto-Gonzalez S, et al. Auscultation of Velcro crackles is associated with usual interstitial pneumonia. Medicine (Baltimore). 2016;95(5):e2573.
Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J. 2012;40(3):519–21.
Boros PW, Enright PL, Quanjer PH, Borsboom GJ, Wesolowski SP, Hyatt RE. Impaired lung compliance and DL,CO but no restrictive ventilatory defect in sarcoidosis. Eur Respir J. 2010;36(6):1315–22.
Diaz-Guzman E, McCarthy K, Siu A, Stoller JK. Frequency and causes of combined obstruction and restriction identified in pulmonary function tests in adults. Respir Care. 2010;55(3):310–6.
Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med. 2002;346(7):484–90.
Jankowich MD, Rounds SIS. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest. 2012;141(1):222–31.
Lin H, Jiang S. Combined pulmonary fibrosis and emphysema (CPFE): an entity different from emphysema or pulmonary fibrosis alone. J Thorac Dis. 2015;7(4):767–79.
Lama VN, Flaherty KR, Toews GB, Colby TV, Travis WD, Long Q, et al. Prognostic value of desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2003;168(9):1084–90.
Kreuter M, Herth FJ, Wacker M, Leidl R, Hellmann A, Pfeifer M, et al. Exploring clinical and epidemiological characteristics of interstitial lung diseases: rationale, aims, and design of a nationwide prospective registry—the EXCITING-ILD Registry. Biomed Res Int. 2015;2015:123876.
Solomon JJ, Olson AL, Fischer A, Bull T, Brown KK, Raghu G. Scleroderma lung disease. Eur Respir Rev. 2013;22(127):6–19.
Gochuico BR, Avila NA, Chow CK, Novero LJ, Wu HP, Ren P, et al. Progressive preclinical interstitial lung disease in rheumatoid arthritis. Arch Intern Med. 2008;168(2):159–66.
Kelly C, Hamilton J. What kills patients with rheumatoid arthritis? Rheumatology (Oxford). 2007;46(2):183–4.
Elhai M, Meune C, Avouac J, Kahan A, Allanore Y. Trends in mortality in patients with systemic sclerosis over 40 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford). 2012;51(6):1017–26.
Homma Y, Ohtsuka Y, Tanimura K, Kusaka H, Munakata M, Kawakami Y, et al. Can interstitial pneumonia as the sole presentation of collagen vascular diseases be differentiated from idiopathic interstitial pneumonia? Respiration. 1995;62(5):248–51.
Fischer A, West SG, Swigris JJ, Brown KK, du Bois RM. Connective tissue disease-associated interstitial lung disease: a call for clarification. Chest. 2010;138(2):251–6.
•• Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44–68 Latest guidelines on the diagnosis of IPF.
Bahmer T, Romagnoli M, Girelli F, Claussen M, Rabe KF. The use of auto-antibody testing in the evaluation of interstitial lung disease (ILD)—a practical approach for the pulmonologist. Respir Med. 2016;113:80–92.
Flaherty KR, Toews GB, Travis WD, Colby TV, Kazerooni EA, Gross BH, et al. Clinical significance of histological classification of idiopathic interstitial pneumonia. Eur Respir J. 2002;19(2):275–83.
Mayo JR. CT evaluation of diffuse infiltrative lung disease: dose considerations and optimal technique. J Thorac Imaging. 2009;24(4):252–9.
Bankier AA, O'Donnell CR, Boiselle PM. Quality initiatives. Respiratory instructions for CT examinations of the lungs: a hands-on guide. Radiographics. 2008;28(4):919–31.
Remy-Jardin M, Campistron P, Amara A, Mastora I, Tillie-Leblond I, Delannoy V, et al. Usefulness of coronal reformations in the diagnostic evaluation of infiltrative lung disease. J Comput Assist Tomogr. 2003;27(2):266–73.
Tokura S, Okuma T, Akira M, Arai T, Inoue Y, Kitaichi M. Utility of expiratory thin-section CT for fibrotic interstitial pneumonia. Acta Radiol. 2014;55(9):1050–5.
Andrade J, Schwarz M, Collard HR, Gentry-Bumpass T, Colby T, Lynch D, et al. The idiopathic pulmonary fibrosis clinical research network (IPFnet): diagnostic and adjudication processes. Chest. 2015;148(4):1034–42.
Hansell DM. Thin-section CT of the lungs: the Hinterland of normal. Radiology. 2010;256(3):695–711.
Kim M, Lee SM, Song JW, Do KH, Lee HJ, Lim S, et al. Added value of prone CT in the assessment of honeycombing and classification of usual interstitial pneumonia pattern. Eur J Radiol. 2017;91:66–70.
Hansell DM, Bankier AA, MacMahon H, McLoud TC, Muller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246(3):697–722.
Jacob J, Hansell DM. HRCT of fibrosing lung disease. Respirology. 2015;20(6):859–72.
Edey AJ, Devaraj AA, Barker RP, Nicholson AG, Wells AU, Hansell DM. Fibrotic idiopathic interstitial pneumonias: HRCT findings that predict mortality. Eur Radiol. 2011;21(8):1586–93.
•• Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138–53 Fleischner Society white paper on the diagnostic criteria for IPF.
Strumpf IJ, Feld MK, Cornelius MJ, Keogh BA, Crystal RG. Safety of fiberoptic bronchoalveolar lavage in evaluation of interstitial lung disease. Chest. 1981;80(3):268–71.
Sakamoto K, Taniguchi H, Kondoh Y, Wakai K, Kimura T, Kataoka K, et al. Acute exacerbation of IPF following diagnostic bronchoalveolar lavage procedures. Respir Med. 2012;106(3):436–42.
• Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185(9):1004–14 Review of the evidence regarding the use of bronchoalveolar lavage for the diagnosis of ILD.
Efared B, Ebang-Atsame G, Rabiou S, Diarra AS, Tahiri L, Hammas N, et al. The diagnostic value of the bronchoalveolar lavage in interstitial lung diseases. J Negat Results Biomed. 2017;16(1):4.
Meyer KC, Raghu G. Bronchoalveolar lavage for the evaluation of interstitial lung disease: is it clinically useful? Eur Respir J. 2011;38(4):761–9.
Dionisio J. Diagnostic flexible bronchoscopy and accessory techniques. Rev Port Pneumol. 2012;18(2):99–106.
Sheth JS, Belperio JA, Fishbein MC, Kazerooni EA, Lagstein A, Murray S, et al. Utility of transbronchial vs surgical lung biopsy in the diagnosis of suspected fibrotic interstitial lung disease. Chest. 2017;151(2):389–99.
Galli JA, Panetta NL, Gaeckle N, Martinez FJ, Moore B, Moore T, et al. Pneumothorax after transbronchial biopsy in pulmonary fibrosis: lessons from the multicenter COMET trial. Lung. 2017;195(5):537–43.
Pankratz DG, Choi Y, Imtiaz U, Fedorowicz GM, Anderson JD, Colby TV, et al. Usual interstitial pneumonia can be detected in transbronchial biopsies using machine learning. Ann Am Thorac Soc. 2017;14(11):1646–54.
Casoni GL, Tomassetti S, Cavazza A, Colby TV, Dubini A, Ryu JH, et al. Transbronchial lung cryobiopsy in the diagnosis of fibrotic interstitial lung diseases. PLoS One. 2014;9(2):e86716.
Tomassetti S, Wells AU, Costabel U, Cavazza A, Colby TV, Rossi G, et al. Bronchoscopic lung cryobiopsy increases diagnostic confidence in the multidisciplinary diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193(7):745–52.
• Johannson KA, Marcoux VS, Ronksley PE, Ryerson CJ. Diagnostic yield and complications of transbronchial lung cryobiopsy for interstitial lung disease. A systematic review and metaanalysis. Ann Am Thorac Soc. 2016;13(10):1828–38 Systematic review on the performance characteristics of transbronchial cryobiopsy for the diagnosis of ILD.
Patel NM, Borczuk AC, Lederer DJ. Cryobiopsy in the diagnosis of interstitial lung disease. A step forward or back? Am J Respir Crit Care Med. 2016;193(7):707–9.
Raj R, Raparia K, Lynch DA, Brown KK. Surgical lung biopsy for interstitial lung diseases. Chest. 2017;151(5):1131–40.
Hutchinson JP, Fogarty AW, McKeever TM, Hubbard RB. In-hospital mortality after surgical lung biopsy for interstitial lung disease in the United States. 2000 to 2011. Am J Respir Crit Care Med. 2016;193(10):1161–7.
Raj R, Brown KK. Mortality related to surgical lung biopsy in patients with interstitial lung disease. The devil is in the denominator. Am J Respir Crit Care Med. 2016;193(10):1082–4.
Flaherty KR, Travis WD, Colby TV, Toews GB, Kazerooni EA, Gross BH, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164(9):1722–7.
Flaherty KR, King TE Jr, Raghu G, Lynch JP 3rd, Colby TV, Travis WD, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med. 2004;170(8):904–10.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Joao de Andrade reports grants from NIH/NHLBI, grants and consulting fee from Genentech and Boehringer Ingelhiem.
Kevin Dsouza declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Interstitial Lung Disease
Rights and permissions
About this article

Cite this article
Dsouza, K., de Andrade, J.A. The Diagnostic Approach to Interstitial Lung Disease. Curr Pulmonol Rep 7, 149–159 (2018). https://doi.org/10.1007/s13665-018-0216-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13665-018-0216-1