
Abstract
The populations of wild honey bee (Apis mellifera) colonies in the USA were decimated after the arrival of a parasitic mite Varroa destructor in the 1980s. However, in some places, wild honey bee colonies survived. In this 3-year study, we analyzed 32 wild and 11 managed colonies in Southwestern Pennsylvania for their maternal genetic ancestries and their levels of Nosema spp. infection. We detected nine mtDNA haplotypes in the 32 wild colonies sampled: six belonged to the Eastern European lineage (C) and three belonged to the Western European lineage (M). We found only three mtDNA haplotypes in the eleven managed colonies sampled, all belonging to the C lineage. Infection levels of N. ceranae were relatively high and fluctuated over time while those of N. apis remained relatively low and constant. There were no differences in N. ceranae or N. apis levels between wild and managed colonies. This study shows that wild honey bee colonies can represent old lineages despite being susceptible to Nosema.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The western honey bee, Apis mellifera, was introduced to North America by European settlers starting in the seventeenth century, mostly for honey production (Nogueira-Neto 1972; Sheppard 1989). Because honey bees are not fully domesticated, swarms quickly escaped from the settler’s hives, moved into the wild, and established a population of wild colonies of “mixed European origin” belonging to the subspecies A. m. carnica, A. m. ligustica, and A. m. mellifera, among others, which spread across the temperate regions of the Americas. Today, wild honey bee colonies living in North America are hybrids of those subspecies that were introduced by the settlers (Moritz et al. 2007). In the USA, the populations of wild honey bee colonies were decimated after the arrival of the ectoparasitic mite Varroa destructor in the 1980s. Beekeepers found that colonies that were not treated for Varroa typically collapsed and died within 2 to 5 years (Korpela et al. 1992; Fries et al. 2006; Boecking and Genersch 2008; Rosenkranz et al. 2010; Dietemann et al. 2013). Thus, it was reasonable to suppose that most, if not all, of the wild colonies in the USA perished because they were left untreated against Varroa mites (Wenner and Bushing 1996; Sanford 2001).
Heavy colony mortality associated with Varroa was indeed reported in the few studies in which populations of wild colonies of honey bees were monitored before, during, and after the arrival of the Varroa mites (e.g., Kraus and Page Jr. 1995; Loper et al. 2006; Villa et al. 2008). One study conducted in the Central Valley of California from 1990 to 1994 found heavy mortality of wild colonies censused before the arrival of Varroa (Kraus and Page Jr. 1995). Another study in Arizona, conducted between 1990 and 1998, showed a precipitous drop in the number of wild colonies after the arrival of tracheal mites (Acarapis woodi), followed by the arrival of Varroa mites (Loper et al. 2006). A longer-term survey conducted from 1991 to 2006 in Louisiana by Villa et al. (2008) monitored the number of swarms (presumed to come from wild colonies) that were captured in bait hives before and after the arrival of V. destructor and found that the capture rate of swarms decreased by 50% after Varroa arrived in 1993. However, within only 5 years, the swarm capture rate recovered to the pre-Varroa levels. Furthermore, the study provided some of the first evidence that in some regions of the USA, wild colonies of honey bees survived and subsequently flourished after the initial decline caused by Varroa.
The most comprehensive studies on feral bees have been conducted in the Arnot Forest in upstate New York. That population of wild colonies was first censused in 1978, which was well before the introduction of V. destructor to North America (Visscher and Seeley, 1982). Upon censusing the population again in 2002 (approximately 10 years after the arrival of Varroa in the area), it was found that the density of wild colonies in the Arnot Forest was the same as before Varroa had arrived there (Seeley 2007). Furthermore, the density of the wild colonies in the Arnot Forest appears to be stable today, as a third survey of this population of wild colonies in 2011 found the same abundance of wild colonies then as in 1978 and 2002 (Seeley 2019).
The persistence of populations of wild colonies of honey bees in North America despite the presence of Varroa shows clearly that wild colonies can be resilient in the presence of pests and pathogens (Szalanski et al. 2016), which could indicate that these populations may harbor enough genetic diversity to select for survivor stock. This is true not just in the USA but also around the world (De Jong and Soares 1997; Le Conte et al. 2007; Moritz et al. 2007; Hinson et al. 2015; Requier et al. 2019). However, Varroa is not the only pathogenic threat to honey bees. A study of a population of wild honey bee colonies living in a protected landscape in South Texas detected low levels of the pathogenic gut microsporidian Nosema ceranae and no evidence of its related species, N. apis (Rangel et al. 2016). A later study examining the effects of urbanization on the health of honey bee colonies across the Eastern US found that the increased pathogen pressure caused by urbanization was ameliorated by a higher expression of immunocompetence genes in wild colonies compared with managed colonies (Youngstead, et al., 2015). These studies demonstrate the importance of studying wild honey bee colonies and their life-history traits across a wide geographical range, so that survivor-stock populations can be identified as potential reservoirs of genetic diversity and resilience to pathogen and parasite pressures.
In this 3-year study, we sampled worker bees from wild colonies living in trees and crevices in and around the Powdermill Nature Reserve in Southwestern Pennsylvania. We also sampled managed colonies living in hives around the reserve to compare the maternal genetic ancestry of the wild and managed colonies in the same area and to begin to compare their health status by determining the prevalence of the pathogens N. ceranae and N. apis. This study provides important information about the health status and diversity of wild honey bee colonies that are living in protected areas in the USA.
2 Materials and methods
2.1 Study site and sample collection
Our study was conducted in the region surrounding the Powdermill Nature Reserve, which is the environmental research station of the Carnegie Museum of Natural History in the Laurel Highlands of Western Pennsylvania (40° 09′ 34.5″ N, 79° 16′ 19.4″ W). The area is mostly rural, with a patchwork of agricultural lands and large tracts of eastern deciduous forest typical of central Appalachia, some of which date from 1900 or earlier.
Nest sites of wild honey bee colonies were either known to one of us (JWW), reported by members of the Westmoreland County Beekeepers, or discovered using the technique of “bee lining” described by Seeley (2016). We defined wild colonies as ones that were living either in natural nest cavities in trees or buildings, or in hives that had been stocked with swarms collected far from beekeepers’ hives, as described by Schiff et al. (1994) and Magnus et al. (2014). We collected foragers coming and going from the entrance with a hand net. To find colonies using the method of bee lining, we captured foragers at flower patches in a special feeder box, allowed the bees to feed ad libitum in the box, and then released them. When the bees returned to the feeder, we labeled them with paint marks, recorded the direction in which they flew home and measured how long they took to make a round trip from the feeder, to their nest, and then back to the feeder. We used the flight direction and “away time” information to estimate the location of the bees’ nest. Then, when several bees were feeding in the feeder box, we closed the box and transported it to a field or clearing in the woods near the suspected location of the bees’ nest. We allowed the labeled bees to re-locate to their new location and then determined their new flight paths home in the same way described above. Once we were close to their home, we started searching for their nest’s entrance (in a tree or building) until we spied bees exiting and entering the hive. We then collected bees from the nest entrance with a net, recorded GPS coordinates for the nest’s location and labeled the tree or building with an identification number.
We collected approximately 100 bees from each nest site and divided the samples such that those destined for pathogen analyses were placed in a − 80 °C freezer until they were transferred to the molecular laboratory on dry ice, and those destined for mitochondrial DNA (mtDNA) analysis were stored in 95% ethanol.
2.2 DNA extraction and mitochondrial DNA analysis
DNA from thoracic muscle was isolated from two individual workers per colony sampled as described in Garnery et al. (1998), with a few modifications. Two bees were sampled from each colony to confirm our results and to check for potential cases of sequencing a bee that drifted from another colony. No cases of bee drift were detected. Briefly, dissection tools were rinsed and cleaned with 10% bleach and 70% ethanol prior to manipulating the bees. Then, each bee was dissected to expose the thorax muscle tissue. This tissue was removed and homogenized using a hand-held homogenizer (BenchTop™ Lab Systems, YG5 3222001). Each homogenization was done on ice in the presence of lysis solution (10 mM EDTA, 100 mM NaCl, 0.1% SDS, 0.25 mg/mL proteinase K) and lasted 30 s. The tissue samples were incubated overnight at 50 °C. Tissue lysate was cleaned with a 1:1 phenol (pH 8): chloroform solution twice, followed by a chloroform extraction. DNA was precipitated with 0.1 volume of 3 M (NaOAc), 2× volume 100% ethanol, and an overnight incubation at − 20 °C. The precipitation was finished with a 30-min centrifugation step (max speed), a wash in 70% ethanol, and then a resuspension in 35 μL of nuclease-free water. DNA was quantified with Qubit dsDNA BR assay (Thermofisher, cat no. Q32853). Working concentrations of 10 ng/μL were used for amplification of the COI-COII mitochondrial region (see below).
2.3 COI-COII amplification
Genetic diversity of the sampled colonies was analyzed in two ways: via digestion of the DraI restriction fragment polymorphism (RFLP) of the cytochrome c oxidase subunit I and II (COI-COII) mitochondrial regions, which identify nucleotide sequences that unique among honey bee mitotypes (Delaney 2008; Delaney et al. 2009; Rortais et al. 2011), or via sequencing. We mitotyped each bee via sequencing using the E2 (F: 5′ GGCAGAATAAGTGCATTG 3′) and H2 (R: 5′ CAATATCATTGA TGACC 3′) primers published by Garnery et al. (1991) to target the COI-COII region of Apis mellifera mitochondria and produce a list of mitotypes. Amplification of the variable region was achieved by using the recommended conditions for OneTaq DNA polymerase (New England Biolabs, Ipswich, MA) for a 25-μL reaction, resulting in a final concentration of 200 μM dNTP, 0.2 μM of each primer, and 1.25 units of OneTaq. The starting template was 20 ng per reaction. Thermal cycler conditions included denaturation and heat activation at 94 °C for 50 s, followed by 30 cycles of denaturation at 94 °C for 15 s, annealing at 48 °C for 20 s, and extension at 68 °C. The final extension was held at 68 °C for 5 min. Successful amplification was confirmed by running 2 μL of the final product via gel electrophoresis (1% agarose, 1× TAE, 80 V for 1 h).
The samples collected in 2014 and 2015 (18 and eleven colonies, respectively) were digested via restriction fragment length polymorphism (RFLP) for mtDNA analysis. To do this, the PCR product was precipitated in 3 M sodium acetate (pH 5.2)/ethanol and then air dried. The final pellet was resuspended in molecular grade water. Digestions (40 μL) with the restriction enzyme DraI (New England Biolabs, Ipswich, MA) were performed at 37 °C overnight.
The mtDNA from samples collected in 2016 (17 colonies) was analyzed via sequencing. To do this, the PCR products were concentrated and cleaned using an E.Z.N.A.® Cycle Pure Kit (Omega Bio-tek, cat no. D6492-02). The samples were forward and reverse sequenced at the Laboratory for Genome Technology (College Station, TX) using ABI Big Dye chemistry. The sequences were aligned and compared with NCBI’s Genbank for published mitotypes, and unique sequences were submitted to GenBank to be archived. The sequence results were also analyzed for DraI cut site.
2.4 DNA extraction and quantitative real-time PCR for Nosema spp. identification
To determine if bees were infected with N. ceranae and/or N. apis, DNA was extracted from ten worker abdomens per colony using the Qiagen DNeasy Blood and Tissue Kit (Germantown, MD, USA). Next, a Nanodrop 2000 Spectrophotometer (Thermo Scientific, Wilmington, DE) was used to assess DNA purity and concentration. Then, qPCR was performed for the simultaneous detection of N. apis and N. ceranae, as described by Traver and Fell (2011). Nosema species–specific TaqMan Gene Expression assays (Applied Biosystems, Foster City, CA, USA) were used to identify the species of Nosema present and to quantify infection levels in each colony sampled. Standard curve quantitation was performed using an Applied Biosystems StepOne Real-Time PCR System to measure the bees’ infection levels, expressed as DNA copy number for each Nosema spp. present in each sample (Traver and Fell 2011).
2.5 Statistical analysis
We performed non-parametric Wilcoxon/Kruskal-Wallis tests to determine whether the average N. ceranae DNA copy number differed significantly between sampling years. We then performed Tukey-Kramer HSD pairwise tests to determine statistical differences between years. We performed Wilcoxon/Kruskal-Wallis tests to establish if N. ceranae DNA copy numbers differed significantly between managed and wild colonies across all 3 years. Because we had mostly zero values for N. apis, we performed a Pearson’s chi-square test to determine if there were differences in N. apis DNA copy numbers across years. We performed non-parametric tests because our data did not meet the assumptions of normality. We used the software JMP 14 Pro (SAS Institute Inc., Cary, NC) for all statistical tests. All descriptive statistics are reported as the mean ± one standard error of the mean (SEM). The level of statistical significance for all tests was set at α = 0.05.
3 Results
We made a total of 47 samplings of bees between 2014 and 2017, 36 from wild colonies and eleven from managed colonies. Specifically, we sampled 18 colonies in 2014, three of which were managed. We sampled eleven colonies in 2015, three of which were managed. Finally, we sampled 17 colonies in 2017, five of which were managed (Figure 1).

Locations of honey bee colonies sampled in and around the Powdermill Nature Reserve, PA, in the summers of 2014 (circles), 2015 (triangles), and 2017 (squares). Wild colonies (white markers) and managed colonies (gray markers) were sampled to determine Nosema ceranae and N. apis DNA copy numbers, as well as mitochondrial DNA haplotypes. Colony 2 was sampled in 2014 and again in 2015. Colonies 8 and 7, which were sampled in 24, and colony 25, which was sampled in 2015, were resampled in 2017. Colonies were labeled in the chronological order in which they were sampled.
3.1 Mitochondrial DNA haplotypes
Overall, we found nine haplotypes represented in the wild colonies, two of which (C1i and M4q) had not been reported previously in GenBank (Table I). Of these, six (66.7%) belonged to the C lineage (A. m. carnica or A. m. ligustica), and three (33.3%) belonged to the M lineage (A. m. mellifera or A. m. iberiensis). Specifically, 85.7% of the wild colonies sampled in 2014 were of the C lineage, while 14.3% were of the M lineage (Figure 2). All the wild colonies sampled in 2015 were of the C lineage. And, in 2017, 83.3% of the wild colonies sampled were of the C lineage, while 16.7% belonged to the M lineage (Figure 2). In contrast, only three haplotypes were represented in the managed colonies across the 3 years, all belonging to the C lineage (Table I, Table S1, Figure 2).

Percent representation of all mitotypes found in wild (a) and managed (b) honey bee colonies sampled around the Powdermill Nature Reserve, PA, in the summers of 2014, 2015, and 2017. The NCBI’s GenBank sequences for all mitotypes are shown in Table II.
Only four of the wild colonies were sampled more than once in different years. Colony 2 was sampled in 2014 and 2015, both times exhibiting the C12 mitotype. Colony 7 was sampled in 2014 and again in 2017, both times exhibiting the newly reported M4q mitotype. Interestingly, colony 8 belonged to the newly reported C1i mitotype in 2014, and then to the C31 mitotype when it was resampled in 2017. And colony 25, which exhibited the C11 mitotype in 2015, had the C1 mitotype in 2017 (Table S1). None of the managed colonies was resampled across years.
3.2 Nosema infection levels
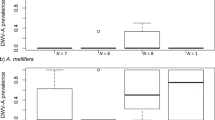
Levels of N. ceranae infection fluctuated over time with 2015 having the highest levels and highest prevalence (Table II, Figure 3). In 2014, 15 of the 18 colonies sampled (83.3%) had bees that tested positive for N. ceranae infection. In 2015, all eleven of the colonies sampled (100%) had bees that tested positive for N. ceranae infection. And in 2017, 16 of the 17 colonies (94.1%) sampled had bees that tested positive for N. ceranae infection. Mean N. ceranae infection levels ranged from a high of 1.05 × 107 ± 9.27 × 106 average copy number in 2015 to a low of 3.50 × 104 ± 2.72 × 104 average copy number in 2017 (Figure 3, Table II). We found significant differences in N. ceranae DNA copy number among colonies in all 3 years (χ2 = 7.38, P = 0.02). Nosema ceranae levels were higher in colonies sampled in 2015 than in those sampled in 2017 (P = 0.03). We found no significant differences in mean N. ceranae level between wild and managed colonies when we pooled the results across years (χ2 = 0.71, P = 0.40).

Mean DNA copy number (± SEM) of Nosema ceranae (gray bars) and N. apis (black bars) obtained from honey bee colonies sampled around the Powdermill Nature Reserve in 2014, 2015, and 2017. Ten worker abdomens were examined from each colony sampled. Nosema spp. identification was performed through qPCR analysis. Colony 2 (denoted with a “*” symbol) was sampled in 2014 and again in 2015. Colonies 8 and 7, which were sampled in 24, and colony 25, which was sampled in 2015 (denoted with a “*” symbol), were resampled in 2017. Colonies 3, 7, 17, and 41 belonged to the M lineage and the specific haplotype is denoted above each colony’s bar. Data are reported on a logarithmic (base 10) scale.
In contrast, the incidence of N. apis infections remained consistently low across the years, with only two colonies (18.8%) testing positive in 2014, one colony (9%) testing positive in 2015, and five colonies (29.4%) testing positive in 2017 (Figure 2). Nosema apis was only detected as a co-infection in bees that we also infected with N. ceranae, which has been observed previously (Traver and Fell 2011). Nosema apis DNA copy number ranged from a high of 2.04 × 103 ± 4.47 × 102 average copy number in 2014, to a low of 1.12 × 103 ± 3.45 × 102 average copy number in 2017 (Table II). We did not find significant differences in N. apis levels across years (Pearson’s χ2 = 16.89, P = 0.53), or between managed and wild colonies (Pearson’s χ2 = 8.69, P = 0.47). Lastly, although we only had a few colonies exhibiting the M lineage, visually, we did not detect a pattern of different Nosema spp. levels between colonies in the C and M lineages (Figure 3).
4 Discussion
We sampled wild and managed colonies of honey bees living in and around the Powdermill Nature Reserve in Southwestern Pennsylvania, to determine their maternal ancestry and the prevalence of the microsporidian gut pathogens N. ceranae and N. apis. Overall, the colonies sampled had relatively low levels of the gut pathogens N. ceranae and N. apis. It has been shown that when managed colonies receive more manipulations and disturbances, the colonies have higher Nosema spp. levels (Oertel 1967). It has also been shown that managed colonies have higher levels and severity of Nosema spp. infection (Manning et al. 2007) and have a higher N. ceranae infection prevalence compared with wild colonies (Gilliam and Taber 1991; Szalanski et al. 2014), with higher infection levels in colonies located in urban areas compared with rural areas (Youngsteadt et al. 2015). But we found no significant differences in N. ceranae or N. apis levels between wild and managed colonies across 3 years. We recognize, however, that our sample size from managed colonies was limited, but our results agree with the study by Thompson et al. (2014), which found no significant differences in N. apis or N. ceranae levels between managed and wild colonies.
Regarding maternal ancestry, we discovered nine distinct mtDNA haplotypes in the wild colonies (Table II), six belonging to the C lineage and three belonging to the M lineage. Interestingly, two of the haplotypes (C1i and M4q) had not yet been reported in GenBank until this study. The M4q haplotype was observed twice, first in 2014 and again in 2017. In contrast, only three mitotypes belonging to the C lineage were found in the managed colonies. None of the colonies sampled belonged to the African lineage (A), which includes A. m. lamarckii and A. m. scutellate, or A. m. iberiensis.
Our results are consistent with other studies, which have reported that the C and M lineages were present in wild colonies across the USA (Sheppard 1988; Schiff et al. 1994; Schiff and Sheppard 1993, 1995, 1996; Delaney 2008; Delaney et al. 2009). For example, Schiff and Sheppard (1993) found that, of the 422 wild honey bee colonies sampled from “non-Africanized” areas in southern states (AL, GA, LA, MS, NC, NM, OK, SC, and TX), 77% belonged to the C lineage, 22% belonged to the M lineage, and only 1% belonged to the A lineage. A more comprehensive sampling made by the same research group later reported that, of 692 wild colonies sampled, 61% belonged to the C lineage, 36% belonged to the M lineage, and only 1% belonged to the A lineage (Schiff et al. 1994). More recently, Magnus et al. (2014) sampled 247 swarms and wild colonies in twelve states (AR, CA, HI, IN, LA, MO, MS, NE, NM, OK, TX, UT) and found that 83% of colonies belonged to the C lineage, 7% were of the M lineage, and 9% belonged to the Near East and Middle East lineage (O). None of the samples belonged to the A lineage, even though it has been reported previously in wild honey bee populations in Texas (Pinto et al. 2004; Rangel et al. 2016). All these studies have found that most of the wild colonies in the USA are of the C lineage, which is also the most common lineage in managed colonies (Schiff and Sheppard 1996; Delaney et al. 2009) and queen breeding operations (Schiff and Sheppard 1996; Magnus et al. 2011) and the only lineage that we observed in the managed colonies we sampled.
Even though only a few of the nest sites inhabited by wild honey bees were sampled multiple times across years, our results suggest that there is much turnover of colonies at sites across time, often of colonies with different maternal haplotypes. For example, colony 8 belonged to the C1i haplotype in 2014, but when the site was sampled again in 2017, the colony living in that cavity belonged to the C31 haplotype. So, evidently, the original colony had died and then the site was re-occupied by a swarm, but because we could not monitor the site steadily, we did not detect the timing of this turnover event. Likewise, the M4q haplotype was observed in colony 7 in 2014 and was again seen in 2017; however, that site was not inhabited in 2015, so the colony observed in this site in 2017 must have been different from the one observed in 2014, albeit with the same haplotype. Furthermore, one of us (JWW) revisited a few of the sites sampled on later dates to record colony survivorship or mortality through the seasons. Interestingly, all nine sites known to have been occupied by wild colonies before the first sampling in 2014 still had active colonies through 2017 and into the spring of 2018. A late, sudden freeze in April 2018 caused a large mortality event such that, of the nine colonies living in these sites, five (44%) died in late spring that year.
This study demonstrates that the wild colonies and the managed colonies living in our study area do not seem to differ in susceptibility to Nosema. It also shows that wild colonies persist for long periods in our study area nonetheless. The presence of the M lineage, which was absent from managed colonies, represents an unbroken lineage of females going back to before this strain of bee was abandoned from apiculture and has therefore persisted without human supervision for perhaps 150 years. Furthermore, many of the wild colonies had mitotypes not represented in the managed ones, which shows that many of the wild colonies are not simply recent escapees from apiaries.
Wild honey bee colonies that have persisted for extended periods of time seem to have adapted well to pathogens and parasites and other environmental stressors without human intervention (Tucker 1976; Villa et al. 2008; Loper et al. 2006; Seeley 2007, Seeley et al. 2015, Seeley 2019). Given the ongoing health issues faced by managed honey bee populations in the USA and the world (Kulhanek et al. 2017), beekeepers and scientists have become interested in the natural history of wild colonies, particularly the traits of “survivor stocks” that have enabled them to survive and thrive despite the many pests and pathogens that affect them (McNeil 2009a, 2009b; Jacobson 2010; Requier et al. 2019). The populations of wild honey bees living in forested areas in the northeast USA, including this study’s population around the Powdermill Nature Reserve in Southwest Pennsylvania, provide us with reservoirs of survivor stock that harbor genetic diversity and pathogen tolerance from which we can draw traits to be used in future breeding programs.
References
Boecking, O., Genersch, E. (2008) Varroosis—the ongoing crisis in beekeeping. J Verbr Lebensm, 3, 221-228.
Collet, T., Ferreira, K.M., Arias, M.C., Soares, A.E., Del Lama, M.A. (2006) Genetic structure of Africanized honeybee populations (Apis mellifera L.) from Brazil and Uruguay viewed through mitochondrial DNA COI-COII patterns. Heredity 97(5), 329-335.
De Jong D., Soares A.E.E. (1997) An isolated population of Italian bees that has survived Varroa jacobsoni infestation without treatment for over 12 years. Am Bee J 137, 742-745.
Delaney, D.A. (2008) Genetic characterization of U.S. honey bee populations. Ph.D. dissertation, Washington State University, Pullman WA.
Delaney, D.A., Meixner, M.D., Schiff, N.M., Sheppard, W.S. (2009) Genetic characterization of commercial honey bee (Hymenoptera, Apidae) populations in the United States by using mitochondrial and microsatellite markers. Ann Entomol Soc Am 102, 666–673.
Dietemann, V., Nazzi, F., Martin, S.J., Anderson, D.L., Locke, B., Delaplane, K.S., Wauquiez, Q., Tannahill, C., Frey, E., Ziegelmann, B., Rosenkranz, P., Ellis, J.D. (2013) Standard methods for Varroa research. J Apic Res 52, 1-53.
Fries, I., Imdorf, A., Rosenkranz, P. (2006) Survival of mite infested (Varroa destructor) honey bee (Apis mellifera) colonies in a Nordic climate. Apidologie 37, 564–570.
Garnery, L., Vautrin, D., Cornuet, J.M., Solignac, M. (1991) Phylogenetic relationships in the genus Apis inferred from mitochondrial DNA sequence data. Apidologie 22, 87-92.
Garnery, L., P. Franck, E. Baudry, D. Vautrin, J.-M. Cornuet, and M. Solignac. 1998. Genetic biodiversity of the west European honeybee (Apis mellifera mellifera and Apis mellifera iberica). I. Mitochondrial DNA. Genet Sel Evol 30, S31–S47.
Gilliam, M., Taber, S.I. (1991) Diseases, pests, and normal microflora of honeybees, Apis mellifera, from feral colonies. J Invertebr Pathol 58(2), 286–289.
Hinson, E.M., Duncan, M., Lim, J., Arundel, J., Oldroyd, B.P. (2015) The density of feral honey bee (Apis mellifera) colonies in South East Australia is greater in undisturbed than in disturbed habitats. Apidologie 46, 403-413.
Jacobson, S. (2010) Locally adapted, varroa resistant honey bees: ideas from several key studies. Am Bee J 150, 777–781.
Korpela, S., Aarhus, A., Fries, I., Hansen, H. (1992) Varroa jacobsoni Oud. in cold climates: population growth, winter mortality and influence on the survival of honey bee colonies. J Apic Res 31, 157-164.
Kraus, B., Page Jr., R.E. (1995) Effect of Varroa jacobsoni (Mesostigmata: Varroidae) on feral Apis mellifera (Hymenoptera: Apidae) in California. Environ Entomol 24, 1473-1480.
Kulhanek, K., Steinhauer, N., Rennich, K., Caron, D.M., Sagili, R.R., Pettis, J.S., Ellis, J., Wilson, M.E., Wilkes, J.T., Tarpy, D.R., Rose, R., Lee, K., Rangel, J., vanEngelsdorp, D. (2017) A national survey of managed honey bee 2015-2016 annual colony losses in the USA. J Apic Res 56, 328-340.
Le Conte, Y., de Vaublanc, G., Crauser, D., Jeanne, F., Rousselle, J-C.,Bécard, J-M. (2007) Honey bee colonies that have survived Varroa destructor. Apidologie 38(6), 566-572.
Loper, G.M., Sammataro, D., Finley, J., Cole, J. (2006) Feral honey bees in southern Arizona 10 years after Varroa infestation. Am Bee J 146, 521-524.
Magnus, R., Tripodi, A.D., Szalanski, A.L. (2011) Mitochondrial DNA diversity of honey bees, Apis mellifera L. (Hymenoptera: Apidae) from queen breeders in the United States. J Apic Sci 55, 5-14.
Magnus, R., Tripodi, A.D., Szalanski, A.L. (2014) Mitochondrial DNA diversity of honey bees (Apis mellifera) from unmanaged colonies and swarms in the United States. Biochem Genet 52, 245-257.
Manning, R., Manning, K., Lancaster, A., Rutkay, L. (2007) Survey of feral honey bee (Apis mellifera) colonies for Nosema apis in Western Australia. Aust J Exp Agric 47(7), 883-886.
McNeil, M.E.A. (2009a) Next up, the survivors, part I. Am Bee J 149, 251–255.
McNeil M.E.A. (2009b) Next up, the survivors, part II. Am Bee J 149, 353–358.
Moritz, R.F.A., Dietemann, V., Crewe, R.M. (2007) Determining colony densities in wild honeybee populations (Apis mellifera) with linked microsatellite DNA markers. J Insect Conserv 12, 455–459.
Nogueira-Neto, P. (1972) Notas sobre a historia da Apicultura Brasileira, in: J.M.F. De Camargo (Ed.), Manual de Apicultura. Editora Agronomia Ceres, São Paulo, pp 17-32.
Oertel, E. (1967) Colony disturbance and Nosema disease. J Apic Res 6(2), 119-120.
Pinto, M.A., Rubink, W.L., Coulson, R.N., Patton, J.C., Johnston, J.S. (2004) Temporal pattern of Africanization in a feral honeybee population from Texas inferred from mitochondrial DNA. Evolution 58, 1047–1055.
Rangel, J., Giresi M., Pinto, M.A., Baum, K.A., Rubink, W.L., Coulson, R.N., Johnston, J.S. (2016) Africanization of a feral honey bee (Apis mellifera) population in South Texas: does a decade make a difference? Ecol Evol 6(7), 2158-69.
Requier, F., Paillet, Y., Laroche, F., Rutschmann, B., Zhang, J., Lombardi, F., Svoboda, M., Steffan-Dewenter, I. (2019) Contribution of European forests to safeguard wild honeybee populations. Conserv Lett. https://doi.org/10.1111/conl.12693.
Rortais, A., Arnold, G., Alburaki, M., Legout, H., Garnery, L. (2011) Review of the DraI COI-COII test for the conservation of the black honeybee (Apis mellifera mellifera). Conserv Genet Resour 3, 383-391.
Rosenkranz, P., Aumeier, P., Ziegelmann, B. (2010) Biology and control of Varroa destructor. J Invertebr Pathol 103, 96–119.
Sanford, M.T. (2001) Introduction, spread, and economic impact of Varroa mites in North America, in: Webster T.C., Delaplane K.S. (Eds.), Mites of the Honey Bee. Dadant and Sons, Hamilton, pp. 149–162.
Schiff, N.M., Sheppard, W.S. (1993) Mitochondrial DNA evidence for the 19th century introduction of African honey bees into the United States. Experientia 49, 350–352.
Schiff, N.M., Sheppard, W.S. (1995) Genetic analysis of commercial honey bees (Hymenoptera: Apidae) from southeastern US. J Econ Entomol 88, 1216–1220.
Schiff, N.M., Sheppard, W.S. (1996) Genetic differentiation in the queen breeding population of the western United States. Apidologie 27, 77–86.
Schiff, N.M., Sheppard, W.S., Loper, G.M., Shimanuki, H. (1994) Genetic diversity of feral honey bee (Hymenoptera: Apidae) populations in the southern United States. Ann Entomol Soc Am 87, 842–848.
Seeley, T.D. (2007) Honey bees of the Arnot Forest: a population of feral colonies persisting with Varroa destructor in the northeastern United States. Apidologie 38, 19–29.
Seeley, T.D. (2016) Following the Wild Bees: The Craft and Science of Bee Hunting. Princeton University Press: Princeton. 184 pp.
Seeley, T.D. (2019) The Lives of Bees: The Untold Story of the Honey Bee in the Wild. Princeton University Press: Princeton. 353pp.
Seeley, T.D., Tarpy, D.R., Griffin, S.R., Carcione, A., Delaney, D.A. (2015) A survivor population of wild colonies of European honeybees in the northeastern United States: investigating its genetic structure. Apidologie 46, 654–666.
Sheppard, W.S. (1988) Comparative study of enzyme polymorphism in United States and European honey bee (Hymenoptera, Apidae) populations. Ann Entomol Soc Am 81, 886–889.
Sheppard, W.S. (1989) A history of the introduction of honey bee races into the United States. Am Bee J 129, 617-619, 664-667.
Solorzano, C.D., Szalanski, A.L., Kence, M., McKern, J.A., Austin, J.W., Kence, A. (2009) Phylogeography and population genetics of honey bees (Apis mellifera) from Turkey based on COI-COII sequence data. Sociobiology 53, 237-246.
Szalanski, A.L., Magnus, R.M. (2010) Mitochondrial DNA characterization of Africanized honey bees (Apis mellifera L.) populations from the USA. J Apic Res 49, 177-185.
Szalanski, A., Tripodi, A.D., Trammel, C.E. (2014) Molecular detection of Nosema apis and N. ceranae from southwestern and south central USA feral Africanized and European honey bees, Apis mellifera (Hymenoptera: Apidae). Fla Entomol 97(2), 585-589.
Szalanski, A.L., Trammel, C.E., Cleary, D., Tripodi, A.D. (2016) Genetic diversity, pathogens and parasites of feral honey bees and swarms from eight Mississippi counties. Midsouth Entomol 9, 67-76.
Thompson, C.E., Biesmeijer, J.C., Allnutt, T.R., Pietravalle, S., Budge, G.E. (2014) Parasite pressures on feral honey bees (Apis mellifera sp.). PLoS One 9(8), e105164.
Traver, B.E., Fell R.D. (2011) Prevalence and infection intensity of Nosema in honey bee (Apis mellifera L.) colonies in Virginia. J Invertebr Pathol 107(1), 43-49.
Tucker, K.W. (1976) Why import bees from abroad? Gleanings Bee Culture 104, 173.
Villa, J.D., Bustamante, D.M., Dunkley, J.P., Escobar, L.A. (2008) Changes in honey bee (Hymenoptera: Apidae) colony swarming and survival pre- and postarrival of Varroa destructor (Mesostigmata: Varroidae) in Louisiana. Ann Entomol Soc Am 101, 867–871.
Visscher, P.K., Seeley, T.D. (1982) Foraging strategy of honeybee colonies in a temperate deciduous forest. Ecology 63, 1790-1801.
Wenner, A.M., Bushing W.W. (1996) Varroa mite spread in the United States. Bee Culture 124, 342–343.
Youngsteadt, E., Appler, R.H., López-Uribe, M.M., Tarpy, D.R., Frank, S.D. (2015) Urbanization increases pathogen pressure on feral and managed honey bees. PLoS One 10(11), e0142031.
Acknowledgments
We would like to thank the members of the Westmoreland County Beekeepers Association, especially Lisa Laskow, for helping us locate some of the colonies and beekeepers sourced in this study.
Contributions
JR, BT, and JW designed the experiments; JR, BT, TDS, and JW collected and interpreted the data; all performed the experiments and analysis; JR, BT, TDS, and JW wrote and revised the paper.
Funding
This study was made possible in part through funding to JR by the Texas AgriLife Research Hatch Project number TEX09557 and USDA-NIFA award number 2015-67013-23170; to BT by the Penn State Schuylkill Research and Development Grant, the Office of Undergraduate Education Research Funds, the Penn State University Schuylkill Faculty and Student Research Endowment, and the Advisory Board for funding both laboratory supplies and undergraduate student funding; and to JWW by the Richard King Mellow Foundation and the Westmoreland County Beekeepers Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Marina Meixner
Diversité génétique des colonies d’abeilles sauvages et d’élevage ( Apis mellifera ) dans le sud-ouest de la Pennsylvanie, et prévalence des pathogènes intestinaux microsporidiens Nosema ceranae et N. apis .
Apis mellifera / colonies sauvages / abeilles domestiques / haplotype / Nosema apis / Nosema ceranae / agents pathogènes.
Genetische Diversität freilebender und bewirtschafteter Völkern der Honigbienes ( Apis mellifera ) im südwestlichen Pennsylvanien, und die Häufigkeit der darmpathogenen Microsporidien Nosema ceranae und N. apis.
Apis mellifera / wildlebende Völker / Honigbienen / Haplotyp / Nosema apis / Nosema ceranae / Pathogene.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1.
(XLSX 12 kb)
Rights and permissions
About this article

Cite this article
Rangel, J., Traver, B., Stoner, M. et al. Genetic diversity of wild and managed honey bees (Apis mellifera) in Southwestern Pennsylvania, and prevalence of the microsporidian gut pathogens Nosema ceranae and N. apis. Apidologie 51, 802–814 (2020). https://doi.org/10.1007/s13592-020-00762-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-020-00762-5